Download to read offline









![Doolittle & Brunet quotes
▶ On accumulating transposable elements: “There is no
selective advantage to individuals within a species for doing
this, and evolution has no foresight [15]!”
▶ “A function concept embedded in one definitional framework
(SE) was (supposedly) refuted by empirical data based on
quite another (CR).”
▶ “it is the irrelevance of the majority of TEs (at least half of
our own DNA) to fitness at the organismal level that means
that “junk” is likely always to be a reasonable way to refer to
it”
▶ Final sentence: “If lungfishes have junk, or at the least
extraordinarily weakly or diffusely functional DNA in their
genomes, why is it that we think we do not?”
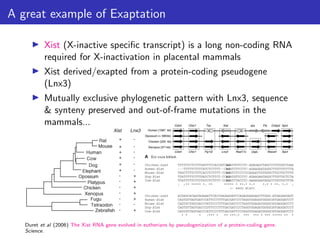
Doolittle & Brunet (2017) On causal roles and selected effects: our genome is mostly junk. BMC Biology.](https://image.slidesharecdn.com/ppgardner-lecture07-genome-function-230710220209-18d277c0/85/ppgardner-lecture07-genome-function-pdf-12-320.jpg)
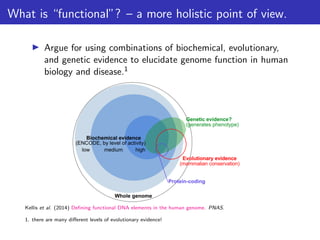












This document outlines a lecture on measuring genome function, exploring the philosophical and biological definitions of 'function' in a genomic context. It discusses various evolutionary concepts, such as selected effects, constructive neutral evolution, and exaptation, while emphasizing the importance of biochemical, evolutionary, and genetic evidence in determining if genomic regions are functional. It also includes references to several studies and provides a framework for self-evaluation exercises in understanding functional DNA elements.






























































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)




