Downloaded 248 times
![[MIT]Introduction to 2GS data analysisDrink faster !June 23, 2011](https://image.slidesharecdn.com/06-23-introductionto2gs-110622202743-phpapp01/85/Introduction-to-second-generation-sequencing-1-320.jpg)
![[MIT]Introduction to 2GS data analysisDrink faster !June 23, 2011](https://image.slidesharecdn.com/06-23-introductionto2gs-110622202743-phpapp01/75/Introduction-to-second-generation-sequencing-1-2048.jpg)
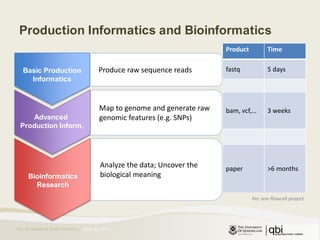
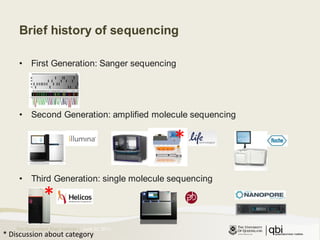

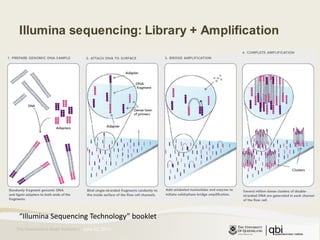

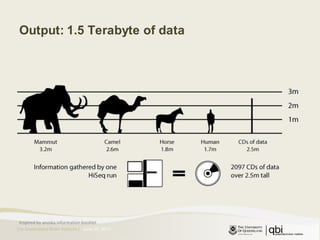
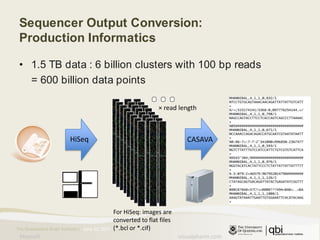

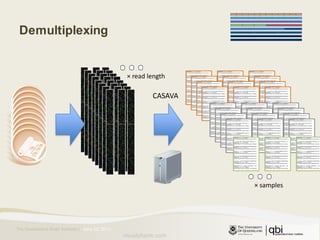

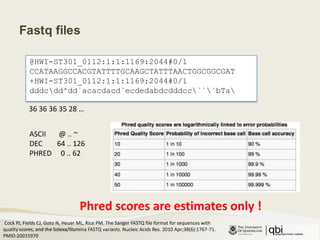
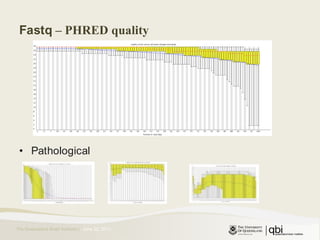




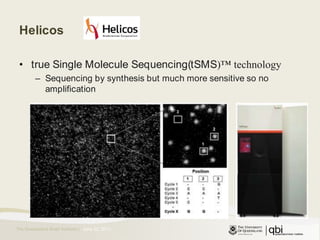

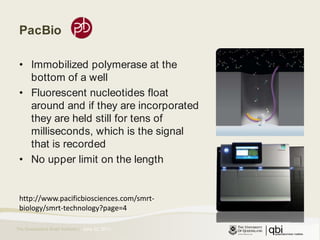


The document provides an overview of various sequencing technologies including Sanger, second generation (amplified molecule), and third generation (single molecule) sequencing. It discusses the data analysis process involved in handling large genomic datasets, specifically focusing on FASTQ file formats and quality control metrics. Additionally, it highlights the bottleneck in modern genomics projects, being primarily at the data analysis stage rather than at data production.









































































































