1. Ref
Reads
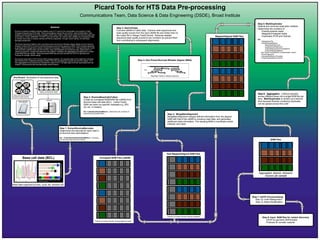
Map/Align FastQ to reference genome
Step 4: Non-Picard Burrows-Wheeler Aligner (BWA)
Step 3: SamToFastq
Converts uBAM to FastQ data. Extracts read sequences and
base quality scores from the input uBAM file and writes them to
the output file in Sanger FastQ format. Reduces adapter
sequence base quality scores to low numbers (to prevent them
from contributing to subsequent alignments).
Aggregated, aligned, deduped,
cleaned per sample
BAM Files
Dye-labeled dideoxynucleotides generate variable
length DNA fragments
Base call data (BCL)
Read data organized by lane, cycle, tile, direction etc.
Pre-Picard: Generation of raw sequence data
Multiplex - Pool libraries and samples
(helps prevent lane-specific artifacts)
Multiplex
Libraries
Prepare multiple libraries per sample.
Libraries have unique barcodes
embedded in adapter sequences
Sequencer
Flow cell lanes
Step 2: IlluminaBaseCallsToSam
Create an unmapped BAM/SAM file (uBAM) from
Illumina base-call data (BCL). Unlike FastQ,
SAM can store run-specific metadata e.g. (RG,
LB, etc. in header).
QC: CollectQualityYieldMetrics - Determines the numbers of
reads that pass quality filters
Step 6: Aggregation - Collects samples
across different lanes into a single BAM file per
lane. MarkDuplicates is carried out a second
time because libraries containing duplicates
can be spread across flow cells
Step 5: MarkDuplicates
Detects and removes duplication artifacts
Determines the numbers of:
-Paired/unpaired reads
-Mapped/unmapped reads
-Duplicates (PCR and Optical)
QC:
ValidateSamFile
CollectMultipleMetrics:
-MeanQualityByCycle
-QualityScoreDistribution
-CollectAlignmentSummaryMetrics
-CollectInsertSizeMetrics
Non-Picard VerifyBamID (Contamination check)
CheckFingerprint
CalculateHsMetrics (Exomes)
CollectGcBiasMetrics (WGS)
Abstract
Picard is a publicly available analysis software suite of 77 tools for the manipulation and analysis of high
throughput sequencing (HTS) data. These manipulations include file conversion, data transformation, data
analysis, and production of an array of quality control (QC) metrics. Data inputs range from Illumina base calls
format (BCL), FastQ, SAM/BAM, VCF/BCF and interval files. QC metrics tools validate and troubleshoot data
at virtually every step of analysis starting with library preparation, through variant calling, and ultimately
genotype assignment.
The Broad Genomics Platform (GP) uses these tools, the Burrows-Wheeler Aligner (BWA) and the Genome
Analysis Toolkit (GATK) in their human whole genome and exome sequence (WGS, WES) analysis pipelines
to genotype and call germline variants. Broad’s GP processes ~4000 exomes and ~1000 genomes per month.
This production pipeline takes raw Illumina BCL data, demultiplexes per sample per lane, aligns them to a
reference sequence, merges this alignment with platform metadata, and aggregates the alignments to obtain
sorted BAM files per sample. These processed BAM files are input directly into GATK for germline and
Firehose for somatic variant calling.
We present tools used in GP’s human WGS analysis pipeline. Our example data is from eight flow cell lanes
that represent four multiplexed samples (8 files). Files are processed per sample per lane from steps 1 to 5,
then aggregated and processed per sample from steps 6 to 8. The eight steps and their associated tools are
outlined, including tools that calculate quality control metrics.
Step 1: ExtractIlluminaBarcodes
Determines the barcode for each read in
an Illumina lane (demultiplex).
QC: CollectIlluminaBasecallingMetrics - Produces
per-lane barcode base-call metrics
Step 7: GATK Pre-processing
Step 7a: Indel Realignment
Step 7b: Base Recalibration
Step 8: Input BAM files for variant discovery
-GATK for germline SNPs/Indels
-Firehose for somatic variants
Grayed out areas indicate missing alignment data
Unmapped BAM Files (uBAM)
Raw Mapped/Aligned SAM Files
Grayed out areas indicate missing metadata
Picard Tools for HTS Data Pre-processing
Communications Team, Data Science & Data Engineering (DSDE), Broad Institute
Mapped/Aligned SAM Files
Step 4: MergeBamAlignment
MergeBamAlignment merges defined information from the aligned
SAM with that of the uBAM to conserve read data, and generates
additional meta information. The resulting BAM is coordinate sorted,
indexed, and clean.