Downloaded 157 times
![[Pink Sherbet Photography]RNAseq analysis: Differential gene expression (2/2)Hopscotch and isoformsAugust 25, 2011](https://image.slidesharecdn.com/08-25-differential-gene-expression-110907205041-phpapp02/85/Differential-gene-expression-1-320.jpg)
![[Pink Sherbet Photography]RNAseq analysis: Differential gene expression (2/2)Hopscotch and isoformsAugust 25, 2011](https://image.slidesharecdn.com/08-25-differential-gene-expression-110907205041-phpapp02/75/Differential-gene-expression-1-2048.jpg)







![Three things to rememberRNAseq captures larger dynamic range (more sensitive)Additional information compared to arrays (e.g. isoforms)Need to make assumptions/compromises (quantification, few replicates) August 25, 2011[cabbit]](https://image.slidesharecdn.com/08-25-differential-gene-expression-110907205041-phpapp02/85/Differential-gene-expression-10-320.jpg)


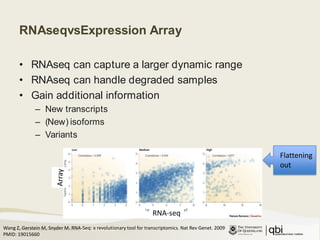
The document outlines RNA sequencing (RNA-seq) analysis methods for differential gene expression, focusing on transcript assembly, quantification, and challenges in data interpretation. It emphasizes RNA-seq's advantages over expression arrays, such as capturing a larger dynamic range and additional information about isoforms. Key challenges include read bias, variability in measurements, and the need for assumptions in quantification methods.










































































