Downloaded 279 times



















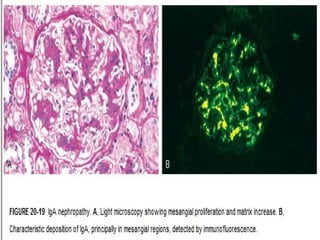








1. IgA nephropathy (Berger's disease) is characterized by prominent IgA deposits in the mesangial regions of the kidney, detected by immunofluorescence microscopy. 2. The disease is caused by increased production of IgA immune complexes that are trapped in the mesangium, where they activate the alternative complement pathway and initiate glomerular injury. 3. Clinical features include recurrent episodes of gross or microscopic hematuria, often following respiratory or gastrointestinal infections. Most patients have normal renal function for decades, but 15-40% may progress to chronic renal failure over 20 years.











































































































