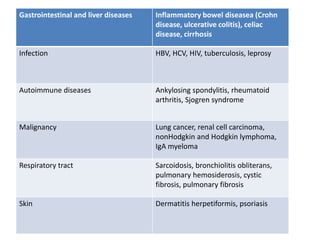
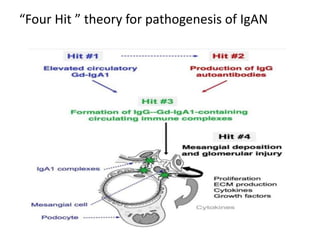
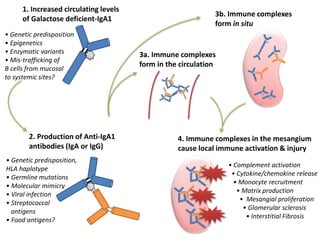
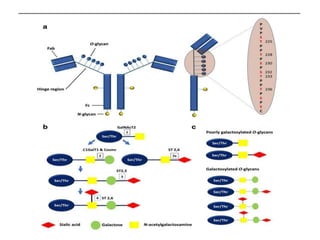
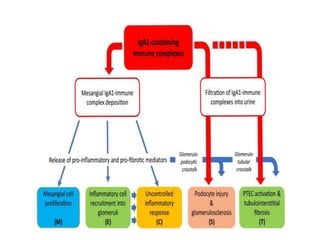
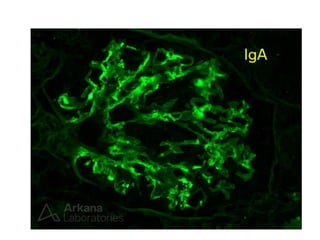
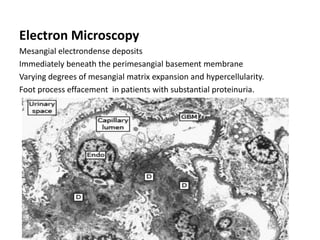
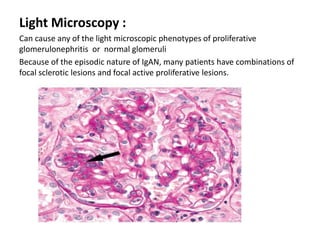
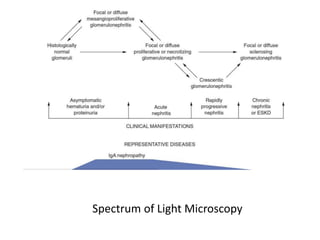
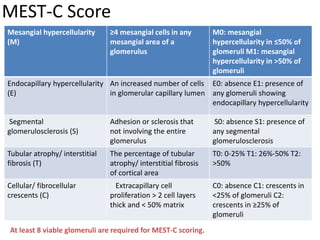
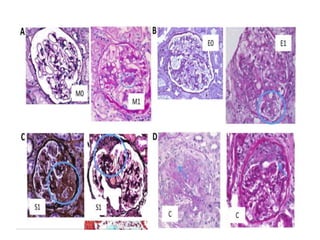
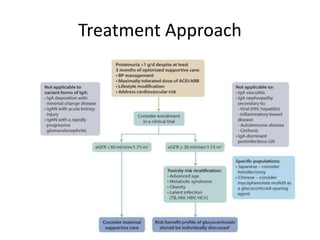
IgA nephropathy, first identified in 1968, is the most common primary glomerulonephritis worldwide and can progress to end-stage kidney disease in a significant number of cases. The pathogenesis involves the deposition of galactose-deficient IgA1 in the mesangium, leading to immune activation and kidney injury, with genetic and environmental factors also playing a role. Diagnosis typically involves renal biopsy, and treatment guidelines suggest evaluating for secondary causes and initiating glucocorticoid therapy for patients with significant proteinuria.