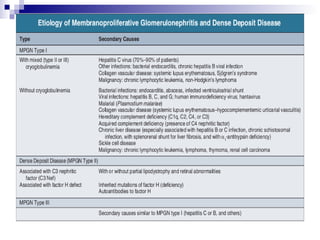
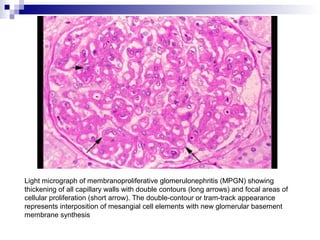
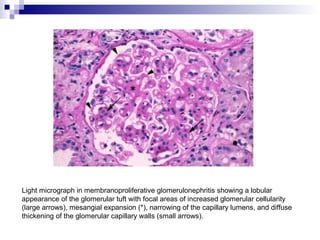
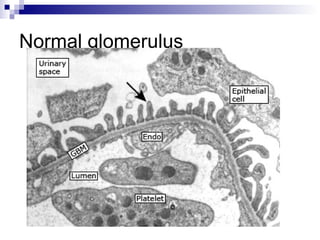
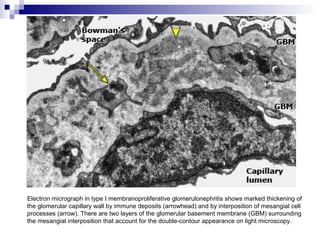
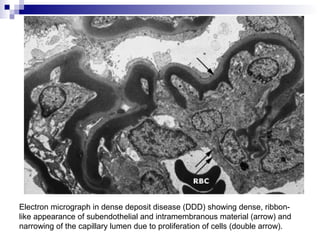
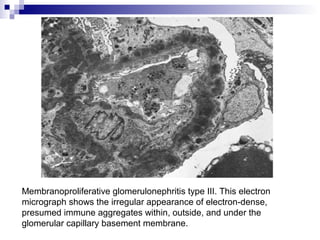
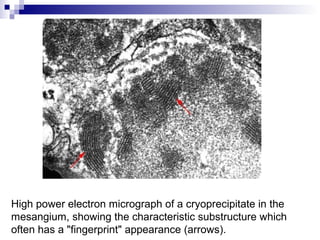
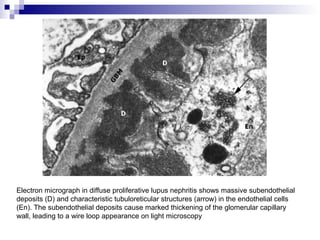
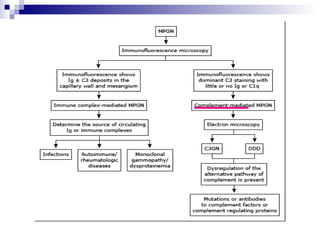
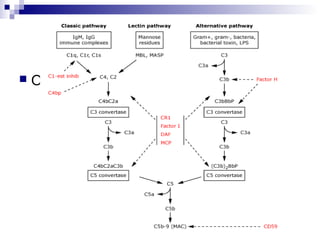
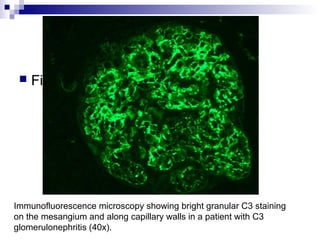
This document provides information on membranoproliferative glomerulonephritis (MPGN). It discusses the histological characteristics of MPGN including thickening of the glomerular basement membrane and increased mesangial and endocapillary cellularity. It presents a new classification system for MPGN that is based on pathogenesis, categorizing it as immune complex-mediated, complement-mediated, or other rare causes. Immune complex-mediated MPGN can result from infections, autoimmune diseases, or monoclonal gammopathies. Complement-mediated MPGN includes dense deposit disease and C3 glomerulonephritis, which are caused by dysregulation of the alternative complement pathway.
































































































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)









![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)





