The document discusses the International Conference on Harmonization (ICH) and its role in standardizing pharmaceutical regulations across Europe, Japan, and the USA to promote public health through timely access to safe and effective drugs. It outlines the objectives and organizational structure of ICH, detailing specific guidelines for quality, safety, and efficacy in drug development, as well as the importance of minimizing redundancy in clinical trials. Various categories of ICH guidelines are provided, along with specific recommendations for testing and evaluating drug substances to ensure their quality and safety.
PRESENTED BY –
PATILABHISHEK SHARAD
DEPARTMENT OF PHARMACEUTICS
RAJARAMBAPU COLLEGE OF PHARMACY, KASEGAON , SANGLI
ICH [ Q ] Guidelines
2.
INTRODUCTION
ICH standsfor “International Conference on Harmonization of
Technical Requirements for Registration of Pharmaceuticals for
Human Use”.
Which is international non-profit Association ,which is unique in
bringing together the regulatory authorities and pharmaceutical
industries.
Where European Union, Japan and the USA involve in scientific
and technical discussions of the testing procedures required to
assess and ensure the safety, quality and efficacy of medicines.
These are the three pillars on which the health of the patients
depend.
3.
ICH Guidelinesaccepted as law in several Countries to ensure
and access the Q,S,E of medicines but are only used as guidance
for the U.S Food and Drug Administration.
Each regulatory co-sponsor implements the guidelines
according to its National or Regional requirements.
They are intended to be used in combination with any regional
requirements.
4.
OBJECTIVES
Promote public healthby early availability of drug in the market.
Maintaining safeguards on quality, safety and efficacy.
Improve efficiency of new drug development ,Reduce registration
cost.
Less expensive drugs for patients.
Prevent the duplication of clinical trails in humans.
Minimize the animal use with out compromising in safety ,efficacy
of the product.
Mutual acceptance of clinical data by regulatory authority.
Reducing testing duplication.
5.
AIM
Many time-consumingand expensive test procedures, in order to
market new products, internationally.
Over rising costs of health care making safe and efficacious new
treatments available to patients in need.
Divergence in technical requirements from country to country.
More economical use of human, animal, and material resources.
Elimination of unnecessary delay in the global development &
availability of new medicines.
Maintaining safeguards on Quality, safety, efficacy and regulatory
obligations to protect public health.
6.
ICH ORGANISATION STRUCTURE
ICH structure consists :
ICH Steering Committee.
ICH Coordinators.
ICH Secretariat.
ICH Working Groups.
The ICH Global Cooperation Group (GCG) and the ICH
MedDRA Management Board are subcommittees of the ICH
Steering Committee.
7.
ICH MEMBERS
EuropeanCommission - European Union (EU).
European Federation of Pharmaceutical Industries and
Associations (EFPIA).
Ministry of Health, Labour and Welfare, Japan (MHLW).
Japan Pharmaceutical Manufacturers Association (JPMA).
US Food and Drug Administration (FDA).
Pharmaceutical Research and Manufacturers of America (PhRMA).
8.
ICH VARIOUS GUIDELINES
ICHhas developed over 50 harmonised Guidelines aimed at eliminating
duplication in the development and registration process, so that a single set
of studies can be generated to demonstrate the quality, safety and efficacy of
a new medicinal product.
Quality-14 Guidelines.
Safety -14 Guidelines.
Efficacy -20 Guidelines.
Multidisciplinary -5 Guidelines.
Electronic Standards for the Transfer of Regulatory Information (ESTRI,
E2B).
Common Technical Document (CTD & eCTD).
Medical dictionary for adverse event reporting and coding of clinical trial
data (MedDRA).
9.
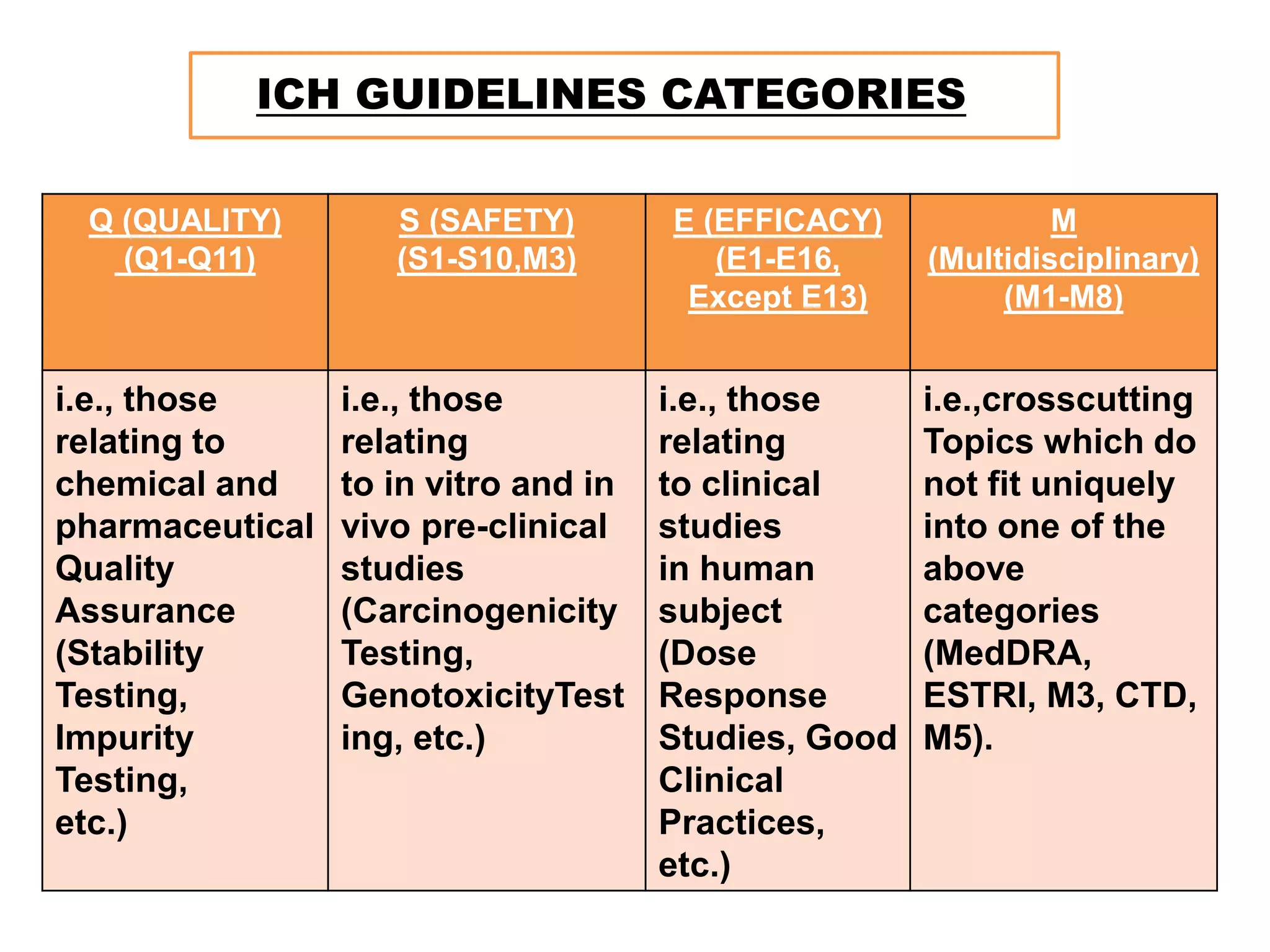
ICH GUIDELINES CATEGORIES
Q(QUALITY)
(Q1-Q11)
S (SAFETY)
(S1-S10,M3)
E (EFFICACY)
(E1-E16,
Except E13)
M
(Multidisciplinary)
(M1-M8)
i.e., those
relating to
chemical and
pharmaceutical
Quality
Assurance
(Stability
Testing,
Impurity
Testing,
etc.)
i.e., those
relating
to in vitro and in
vivo pre-clinical
studies
(Carcinogenicity
Testing,
GenotoxicityTest
ing, etc.)
i.e., those
relating
to clinical
studies
in human
subject
(Dose
Response
Studies, Good
Clinical
Practices,
etc.)
i.e.,crosscutting
Topics which do
not fit uniquely
into one of the
above
categories
(MedDRA,
ESTRI, M3, CTD,
M5).
10.
[Q] QUALITY GUIDELINES
Q1A - Q1F :Stability
Q2 : Analytical Validation
Q3A - Q3D : Impurities
Q4 - Q4B : Pharmacopoeias
Q5A - Q5E : Quality of Biotechnological Products
Q6A- Q6B : Specifications
Q7 : Good Manufacturing Practice
Q8 : Pharmaceutical Development
Q9 : Quality Risk Management
Q10 : Pharmaceutical Quality System
Q11 : Development and Manufacture of Drug Substances
Q12 : Lifecycle Management
Q13 : Continuous Manufacturing of Drug Substances and Drug
Products
Q14 : Analytical Procedure Development
11.
Q1(STABILITY)
Q1A-STABILITY TESTING OFNEW DRUG SUBSTANCES AND
PRODUCTS, The purpose of stability testing is to provide evidence
on how the quality of a drug substance or drug product varies with
time under the influence of a variety of environmental factors such as
temperature, humidity, and light, and to establish a re-test period for
the drug substance or a shelf life for the drug product and
recommended storage conditions.
GENERAL CASE
13.
Q1B--PHOTOSTABILITY TESTING OFNEW DRUG
SUBSTANCES AND PRODUCTS
A systematic approach to photostability testing is
recommended covering, as appropriate, studies such as:
i) Tests on the drug substance.
ii) Tests on the exposed drug product outside of the immediate
pack.
iii) Tests on the drug product in the immediate pack and if
necessary.
iv) Tests on the drug product in the marketing pack.
14.
PROCEDURE -
Samples shouldbe exposed to
light providing an overall
illumination of not less than 1.2
million lum. hours and an
integrated near ultraviolet energy
of not less than 200 watt
hours/square meter to allow direct
comparisons to be
made between the drug substance
and drug product
And after exposure to light the
absorbance of sample is taken in
UV visible spectrophotometry to
know the actual content of
drug after degradation by light and
after that decision is made,
either the product is photosensitive
or not.
15.
Q1C- Stability Testingfor New Drugs and
Products
NEW DOSAGE FORMS
A new dosage form is defined as a drug product which is a different
pharmaceutical product type, but contains the same active substance
as included in the existing drug product approved by the pertinent
regulatory authority.
Such pharmaceutical product types include products of different
administration route (e.g., oral to parenteral), new specific
functionality/delivery systems (e.g., immediate release tablet to
modified release tablet) and different dosage forms of the same
administration route (e.g., capsule to tablet, solution to suspension).
Stability protocols for new dosage forms should follow the
guidance in the parent stability guideline in principle. However, a
reduced stability database at submission time (e.g., 6 months
accelerated and 6 months long term data from ongoing studies) may
be acceptable in certain justified cases.
16.
Q1D: Bracketingand Matrixing Designs for Stability Testing of
New Drug Substances and Products.
Q1E: Evaluation of Stability Data This guideline addresses the
evaluation of stability data that should be submitted in registration
applications for new molecular entities and associated drug
products. The guideline provides recommendations on establishing
shelf lives for drug substances and drug products intended for
storage at or below “room temperature”.
Q1F: Stability Data Package for Registration Applications in
Climatic Zones III and IV Describes harmonized global stability
testing requirements in order to facilitate access to medicines by
reducing the number of different storage conditions. WHO
conducted a survey amongst their member states to find consensus
on 30°C/65% RH as the long term storage conditions for hot-dry and
hot-humid regions.
17.
Q2(R1): VALIDATION OFANALYTICAL PROCEDURES;
TEXT AND METHODOLOGY
Types of Analytical Procedures to be
Validated :
- Identification tests
- Quantitative tests for impurities'
content
- Limit tests for the control of
impurities
- Quantitative tests of the active
moiety in samples of drug substance
or drug product or other selected
component(s) in the drug product.
Typical validation characteristics are;
o Accuracy
o Precision
o Repeatability
o Intermediate Precision
o Specificity Detection Limit
o Quantitation Limit
o Linearity
o Range
Furthermore revalidation may be
necessaryin the following
circumstances: -
-changes in the synthesis of the
drug substance
- changes in the composition of
the finished product
-changes in the analytical
procedure.
The degree of revalidation
required depends on the nature
of the changes.
Certain other changes may
requirevalidation as well.
18.
Q3A-IMPURITIES IN NEWDRUG SUBSTANCES
This document is intended to provide guidance for registration
applications on the content and qualification of impurities in new
drug substances produced by chemical synthesis and not
previously registered in a region or member state.
The following types of drug substances are not covered in this
guideline:
Biological/biotechnological, peptide, oligonucleotide,
radiopharmaceutical, fermentation product and semi-synthetic
products derived therefrom, herbal products, and crude products of
animal or plant origin.
Impurities can be classified into the following categories:
Organic impurities (process- and drug-related)
Inorganic impurities
Residual solvents
19.
Q3B-IMPURITIES IN NEWDRUG PRODUCTS
This document provides guidance for registration
applications on the content and qualification of
impurities in new drug products produced from
chemically synthesised new drug substances not
previously registered in a region or member state.
This guideline addresses only those impurities in new
drug products classified as degradation products of the
drug substance or reaction products of the drug
substance with an excipient and/or immediate container
closure system.
20.
Q3C-IMPURITIES: GUIDELINE FORRESIDUAL
SOLVENTS
The objective of this guideline is to recommend acceptable amounts for
residual solvents in
pharmaceuticals for the safety of the patient.
The guideline recommends use of less toxic solvents and describes levels
considered to be
toxicologically acceptable for some residual solvents.
Classification of Residual Solvents by Risk Assessment
Class 1 solvents: Solvents to be avoided
Known human carcinogens, strongly suspected human carcinogens, and
environmental hazards.for ex.
Class 2 solvents: Solvents to be limited
Non-genotoxic animal carcinogens or possible causative agents of other
irreversible toxicity such as neurotoxicity or teratogenicity. Solvents
suspected of other significant but reversible toxicities.
Class 3 solvents: Solvents with low toxic potential
Solvents with low toxic potential to man; no health-based exposure limit is
needed.
Class 3 solvents have PDEs of 50 mg or more per day.
21.
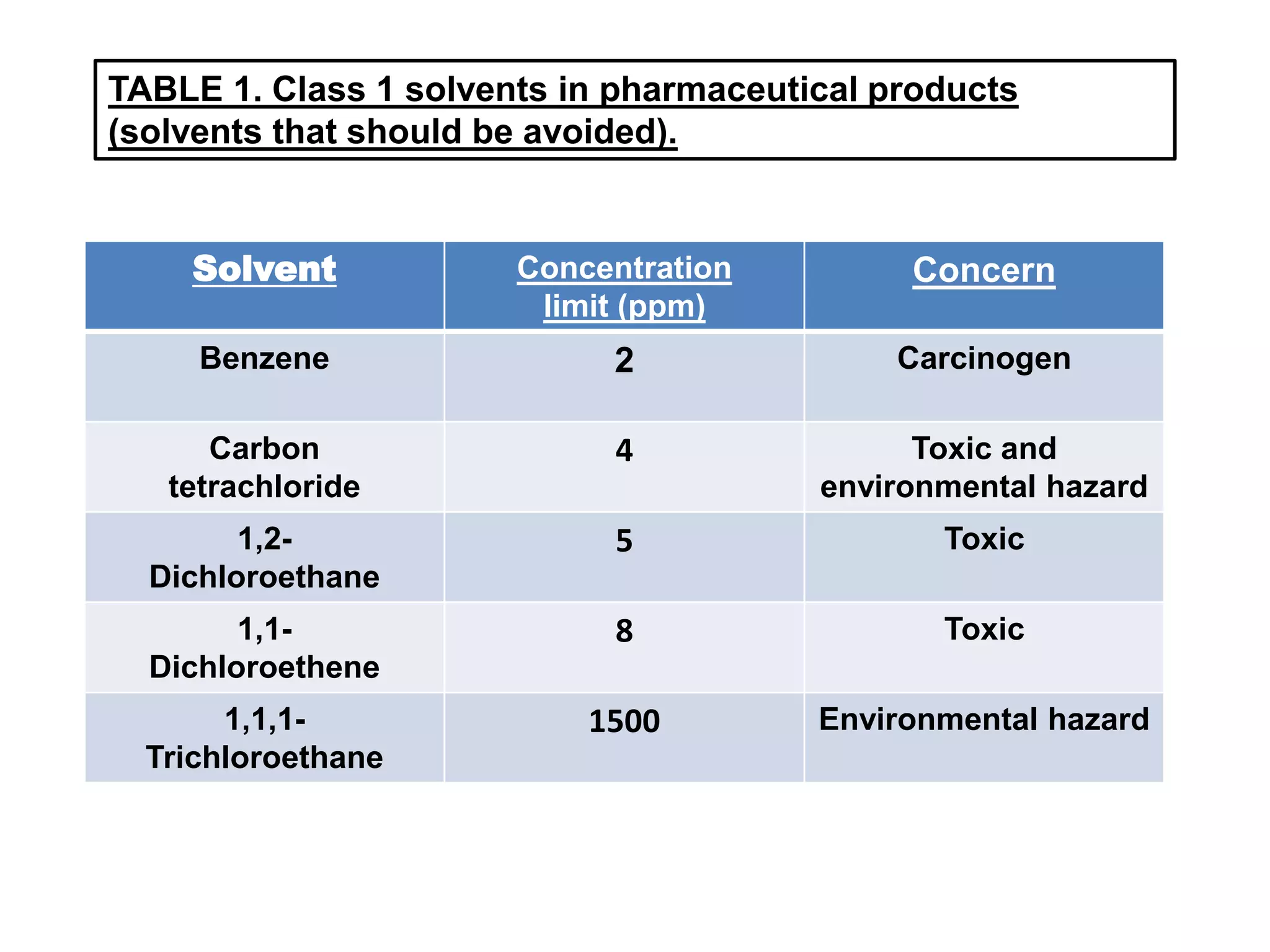
TABLE 1. Class1 solvents in pharmaceutical products
(solvents that should be avoided).
Solvent Concentration
limit (ppm)
Concern
Benzene 2 Carcinogen
Carbon
tetrachloride
4 Toxic and
environmental hazard
1,2-
Dichloroethane
5 Toxic
1,1-
Dichloroethene
8 Toxic
1,1,1-
Trichloroethane
1500 Environmental hazard
22.
Q3D-GUIDELINE FOR ELEMENTALIMPURITIES
Elemental impurities in drug products may arise from several sources;
they may be residual catalysts that were added intentionally in synthesis
or may be present as impurities (e.g., through interactions with
processing equipment or container/closure systems or by being present
in components of the drug product).
Because elemental impurities do not provide any therapeutic benefit to
the patient, their levels in the drug product should be controlled within
acceptable limits.
There are three parts of this guideline:
The evaluation of the toxicity data for potential elemental impurities;
The establishment of a Permitted Daily Exposure (PDE) for each
element of toxicological concern
Application of a risk-based approach to control elemental impurities in
drug products for ex. Cd,Pb,As,Hg,V,Pd,etc.
23.
Q4B-EVALUATION AND RECOMMENDATIONOF
PHARMACOPOEIAL TEXTS FOR USE IN THE ICH REGIONS ON
RESIDUE ON IGNITION/SULPHATED ASH GENERAL CHAPTER
It is further divided into following 14 annex
• Q4B Annex 1: Evaluation and Recommendation of Pharmacopoeial Texts for Use in the
ICH Regions on Residue on Ignition/Sulphated Ash.
• Annex 2:Test for Extractable Volume of Parenteral Preparations
• Annex 3: Test for Particulate Contamination: Sub-Visible Particles
• Annex 4A: Microbiological Examination of Non-Sterile Products: Microbial Enumeration
Tests
• Annex 4B: Microbiological Examination of Non-Sterile Products: Tests for Specified Micro-
organisms
• Annex 4C: Microbiological Examination of Non-Sterile Products: Acceptance Criteria for
Pharmaceutical Preparations and Substances for Pharmaceutical Use
• Annex 5:Disintegration Test
• Annex 6: Uniformity of Dosage Units
• Annex 7: Dissolution Test
• Annex 8: Sterility Test
• Annex 9: Tablet Friability
• Annex 10: Polyacrylamide Gel Electrophoresis
• Annex 11: Capillary Electrophoresis
• Annex 12: Analytical Sieving
• Annex 13: Bulk Density and Tapped Density of Powders
• Annex14 :Bacterial Endotoxins Test
24.
Q5A-Q5E---Quality of BiotechnologicalProducts:
Q5A(R1): Viral Safety Evaluation of Biotechnology Products Derived
from Cell Lines of Human or Animal Origin:
This document is concerned with testing and evaluation of the viral
safety of biotechnology products derived from cell lines of human or
animal origin (i.e. mammalian, avian, insect)
The objective is to provide a general framework for virus testing
experiments for the evaluation of virus clearance and the design of
viral tests and clearance evaluation studies.
Three principal, complementary approaches have evolved to
control the potential viral contamination of Biotechnology products
a) selecting and testing cell lines and other raw materials, including
media components, for the absence of undesirable viruses which
may be infectious and/or pathogenic for humans.
b) Testing the capacity of the processes to clear infectious viruses.
c) testing the product at appropriate steps for absence of
contminating infectious viruses.
25.
Q6 : Specificationsfor New Drug Substances and Products
Bulk drug substance and final product specifications are key parts of the
core documentation for world-wide product license applications.
This leads to conflicting standards for the same product, increased
expenses and opportunities for error as well as a potential cause for
interruption of product supply.
Q6A: Specifications : Test Procedures and Acceptance Criteria for New
Drug Substances and New Drug Products : Chemical Substances
The main objective of this guideline is to establish a single set of global
specifications for new drug substances and new drug products.
This guideline addresses specifications, i.e., those tests, procedures, and
acceptance criteria which play a major role in assuring the quality of the
new drug substance and new drug product during shelf life.
26.
Q7: Good ManufacturingPractice Guide for Active
Pharmaceutical Ingredients
The main objective of this guideline is that to maintain the
quality of the active pharmaceutical ingredients:
Personnel
Buildings and Facilities
Process equipment
Documentation and Records
27.
Q8(R2): Pharmaceutical Development
This guideline is intended to provide guidance on the contents of
Pharmaceutical Development of drug products.
The aim of pharmaceutical development is to design a quality
product and its manufacturing process to consistently deliver the
intended performance of the product.
The Pharmaceutical Development section also describe the type
of dosage form and the formulation that are suitable for the intended
use.
Q8 gives information about Drug Substance, Excipients,
Container Closure System.
28.
Q9: Quality RiskManagement
The purpose of this document is to offer a systematic approach
to quality risk management.
This guideline provides principles and tools for quality risk
management that can be applied to all aspects of pharmaceutical
quality including development, manufacturing, distribution; and
the inspection and submission/review processes throughout the
lifecycle of drug substances and drug (medicinal) products,
biological and biotechnological products, including the use of
raw materials, solvents, excipients, packaging and labeling
materials.
29.
Q10: Pharmaceutical QualitySystem
This document establishes a new ICH tripartite guideline
describing a model for an effective quality management system for
the pharmaceutical industry, referred to as the Pharmaceutical
Quality System.
Comprehensive model for an effective pharmaceutical quality
system is based on International Standards Organization (ISO)
quality concepts, includes applicable Good Manufacturing Practice
(GMP) regulations
30.
Q11: Development andManufacture of Drug Substances:
This new guidance is proposed for Active Pharmaceutical
Ingredients (APIs) harmonising the scientific and technical
principles relating to the description and justification of the
development and manufacturing process (CTD sections S 2.2. - S
2.6) of Drug Substances including both chemical entities and
biotechnological/biological entities.
Q12: Lifecycle Management :
This new guideline is proposed to provide guidance on a
framework to facilitate the management of post-approval
Chemistry, Manufacturing and Controls (CMC) changes in a
more predictable and efficient manner across the product
lifecycle. Adoption of this new ICH Guideline will promote
innovation and continual improvement, and strengthen quality
assurance and reliable supply of product, including proactive
planning of supply chain adjustments.
31.
Q13: Continous manufacturingof drug substances and drug
Products :
This topic was endorsed by the Assembly in June 2018. This new
Guideline is proposed to:
• Capture key technical and regulatory considerations that
promote harmonisation, including certain Current Good
Manufacturing Practices (CGMP) elements specific to Continuous
Manufacturing (CM).
• Allow drug manufacturers to employ flexible approaches to
develop, implement, or integrate CM for the manufacture – drug
substances and drug products – of small molecules and
therapeutic proteins for new and existing products,
• Provide guidance to industry and regulatory agencies regarding
regulatory expectations on the development, implementation, and
assessment of CM technologies used in the manufacture of drug
substances and drug products.
32.
Q 14 Analyticalprocedure development :
The new guideline is proposed to harmonise the
scientific approaches of Analytical Procedure
Development, and to provide the principles relating to
the description of Analytical Procedure Development
process. This new guideline is intended to improve
regulatory communication between industry and
regulators and facilitate more efficient, sound scientific
and risk-based approval as well as post-approval
change management of analytical procedures.
33.
REFERENCES
ICH guidlinesof quality and safety
http://www.ich.org/products/guidelines/quality/article/qualityguidelines.
html
http://www.ich.org/products/guidelines/safety/article/safetyguidelines.
Html
ICH GUIDELINES PRESENTATIONS BY FOLLOWING :
1. PRSENTED BY : MANISH SHANKARPURE
M.PHARM ( QUALITY ASSURANCES AND TECHNIQUES )
2. BASHANT KUMAR SAH
Mpharm 1st pharmaceutics
Nargund college of pharmacy
3. JAYA PRAKASH V
REGULATORY AFFAIRS
REG NO: 218311
![PRESENTED BY –
PATIL ABHISHEK SHARAD
DEPARTMENT OF PHARMACEUTICS
RAJARAMBAPU COLLEGE OF PHARMACY, KASEGAON , SANGLI
ICH [ Q ] Guidelines](https://image.slidesharecdn.com/ichabhishek-210812054107/75/ICH-Q-Guidelines-1-2048.jpg)







![[Q] QUALITY GUIDELINES
Q1A - Q1F :Stability
Q2 : Analytical Validation
Q3A - Q3D : Impurities
Q4 - Q4B : Pharmacopoeias
Q5A - Q5E : Quality of Biotechnological Products
Q6A- Q6B : Specifications
Q7 : Good Manufacturing Practice
Q8 : Pharmaceutical Development
Q9 : Quality Risk Management
Q10 : Pharmaceutical Quality System
Q11 : Development and Manufacture of Drug Substances
Q12 : Lifecycle Management
Q13 : Continuous Manufacturing of Drug Substances and Drug
Products
Q14 : Analytical Procedure Development](https://image.slidesharecdn.com/ichabhishek-210812054107/75/ICH-Q-Guidelines-10-2048.jpg)






















![ICH [ Q ] Guidelines](https://image.slidesharecdn.com/ichabhishek-210812054107/75/ICH-Q-Guidelines-34-2048.jpg)



































































