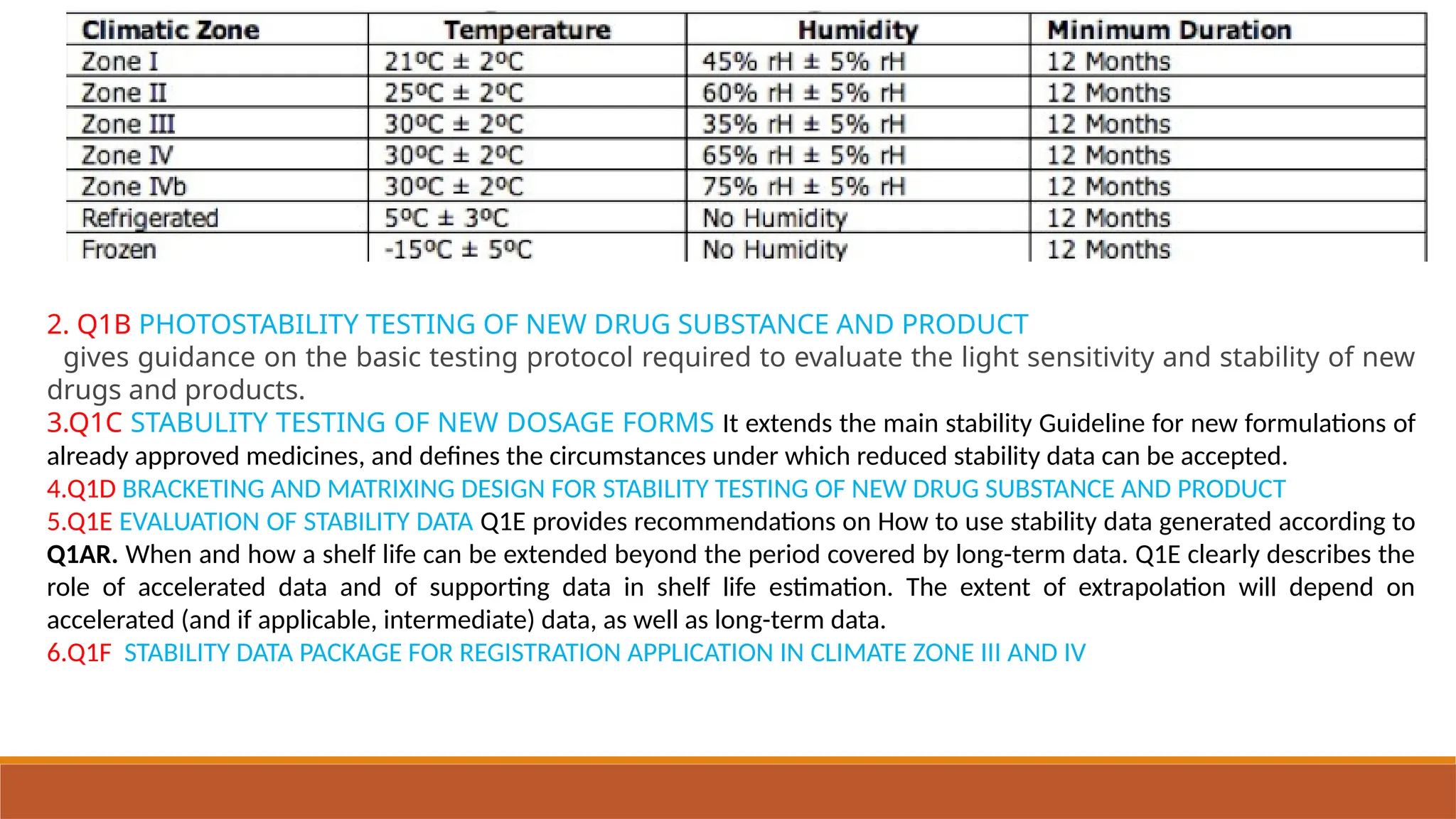
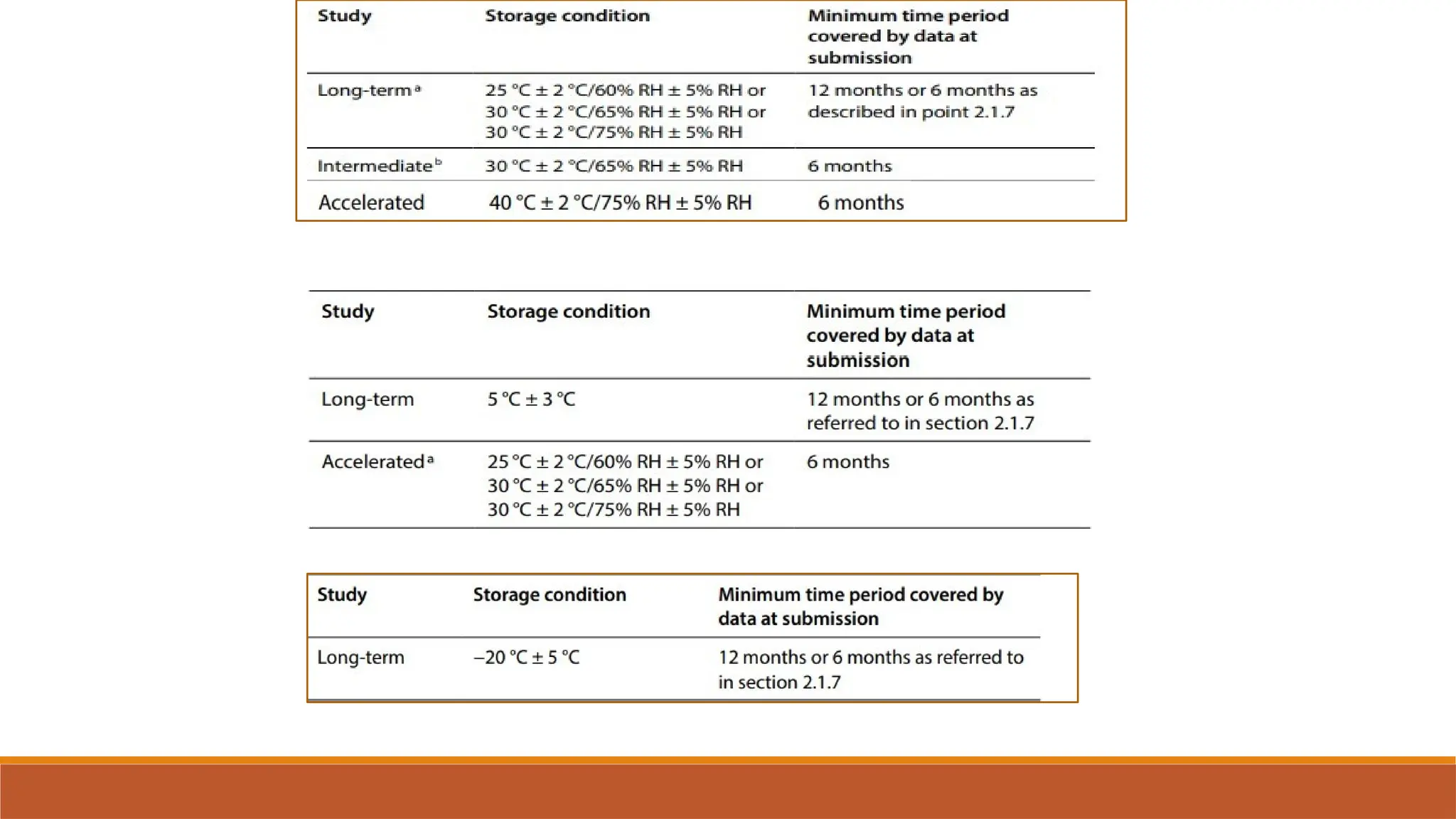
The document outlines the International Council for Harmonisation (ICH) guidelines for drug product evaluation and registration, emphasizing the importance of independent evaluations driven by past tragedies. It details various quality guidelines across different Q-series addressing stability testing, impurities, and manufacturing practices to ensure the safety and efficacy of medicinal products globally. The document highlights ICH's mission to harmonize international regulations for pharmaceutical industry efficiency and product quality.
















![ICH [ Q ] Guidelines](https://cdn.slidesharecdn.com/ss_thumbnails/ichabhishek-210812054107-thumbnail.jpg?width=640&height=640&fit=bounds)
























![Apporach to lung biopsy [Auto-saved].pptx latest](https://cdn.slidesharecdn.com/ss_thumbnails/apporachtolungbiopsyauto-saved-251211225655-93258539-thumbnail.jpg?width=640&height=640&fit=bounds)
















