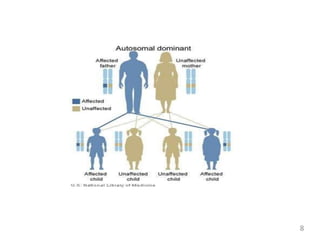
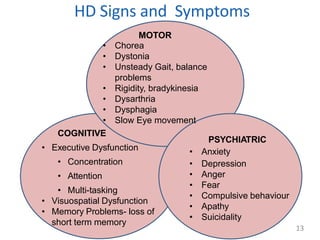
Huntington's disease is an autosomal dominant neurodegenerative disorder characterized by motor, cognitive, and psychiatric symptoms, usually manifesting in adulthood. The faulty gene responsible for the disease was identified in 1993, and those affected have a 50% chance of passing it on to their offspring. While there is currently no cure, treatment focuses on symptom management through medications and supportive therapies.























































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)


















