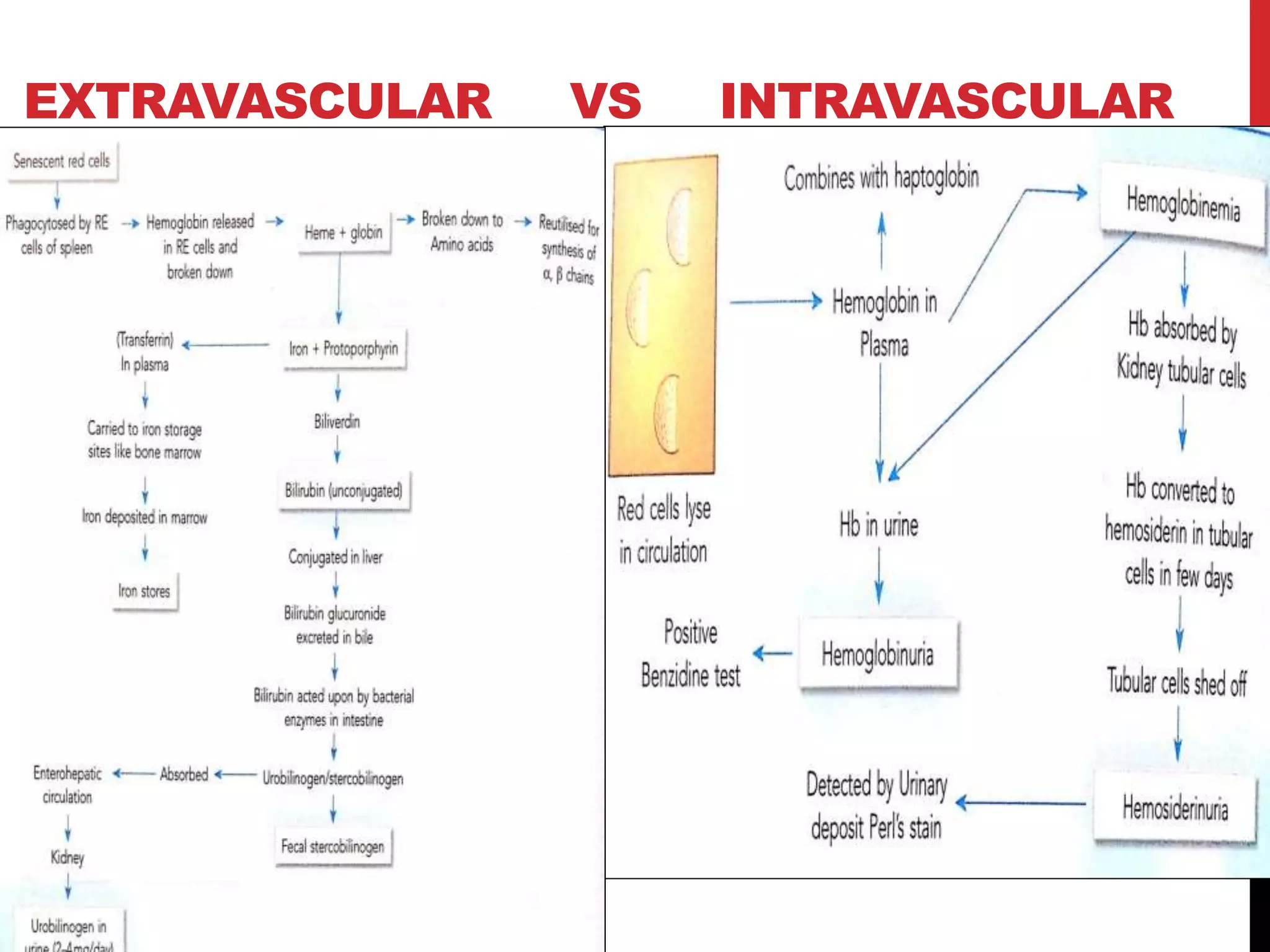
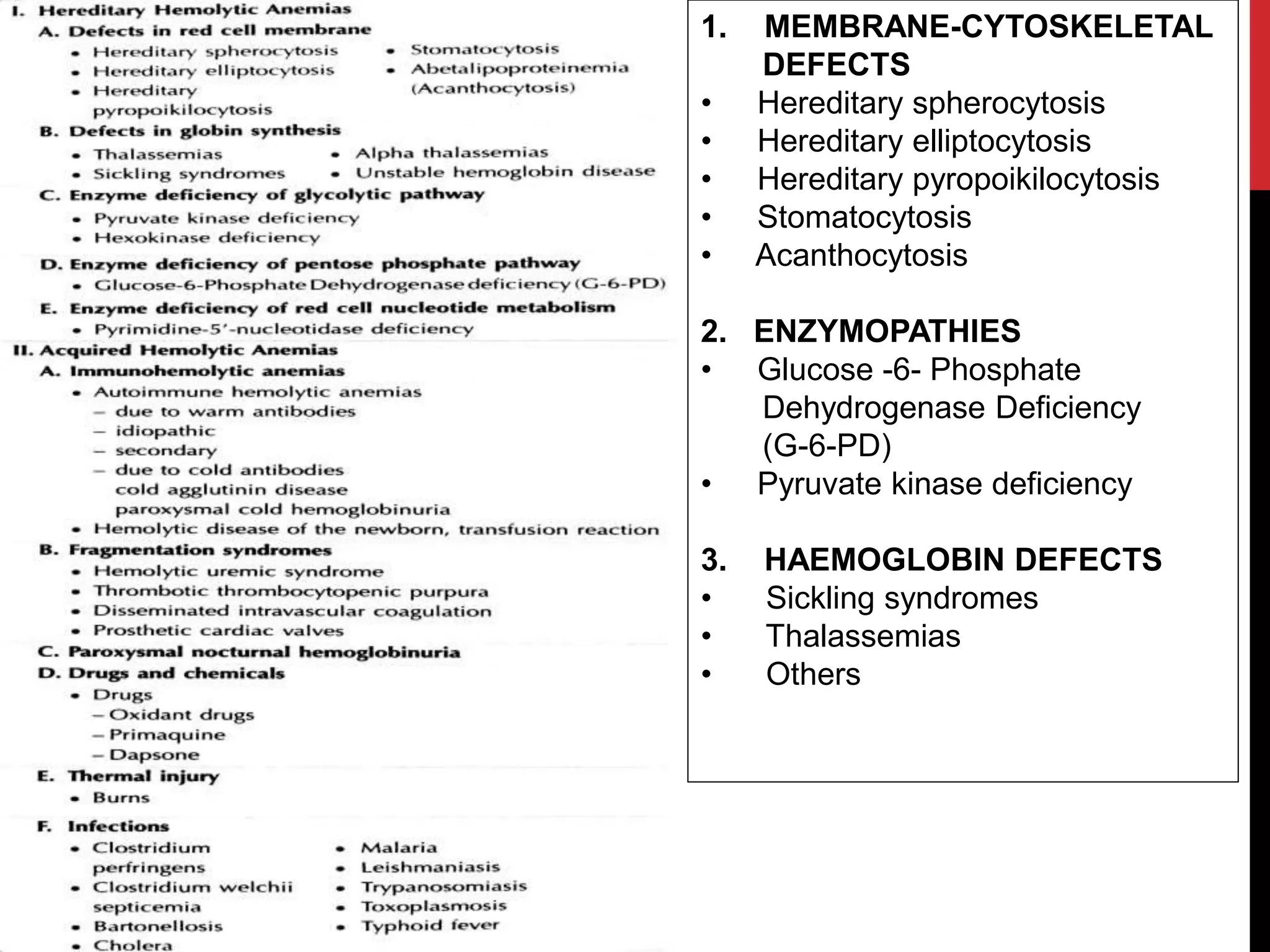
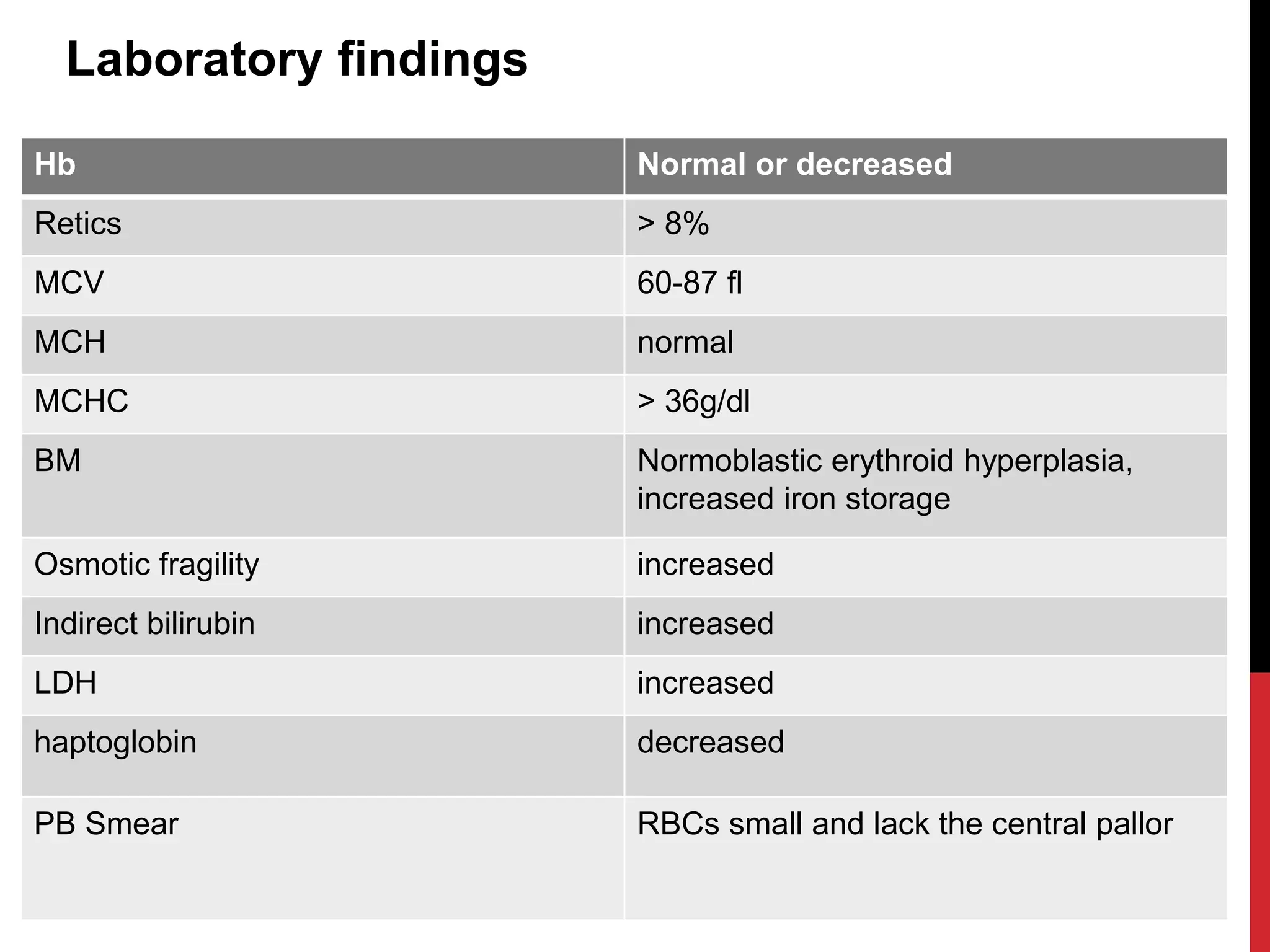
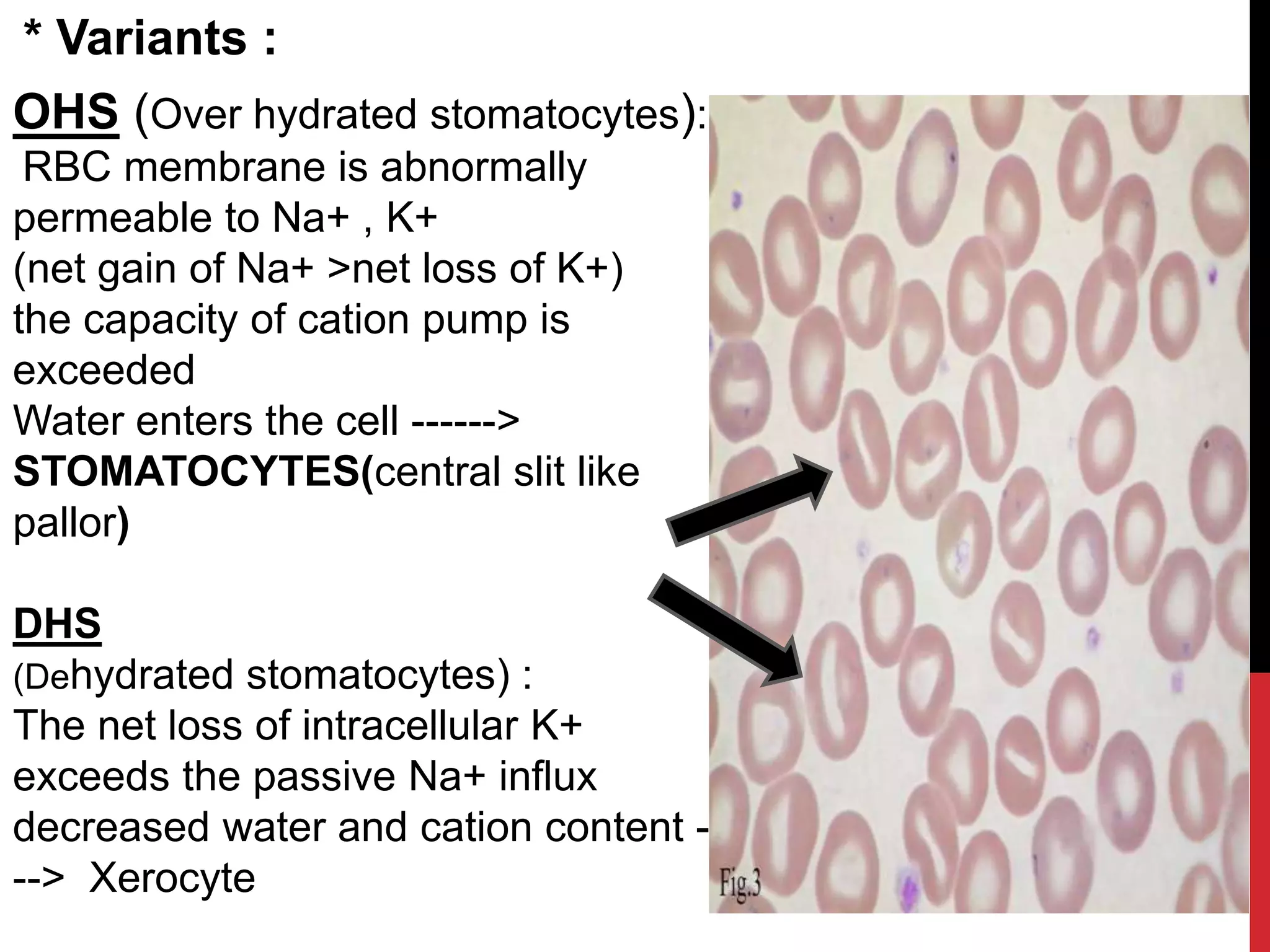
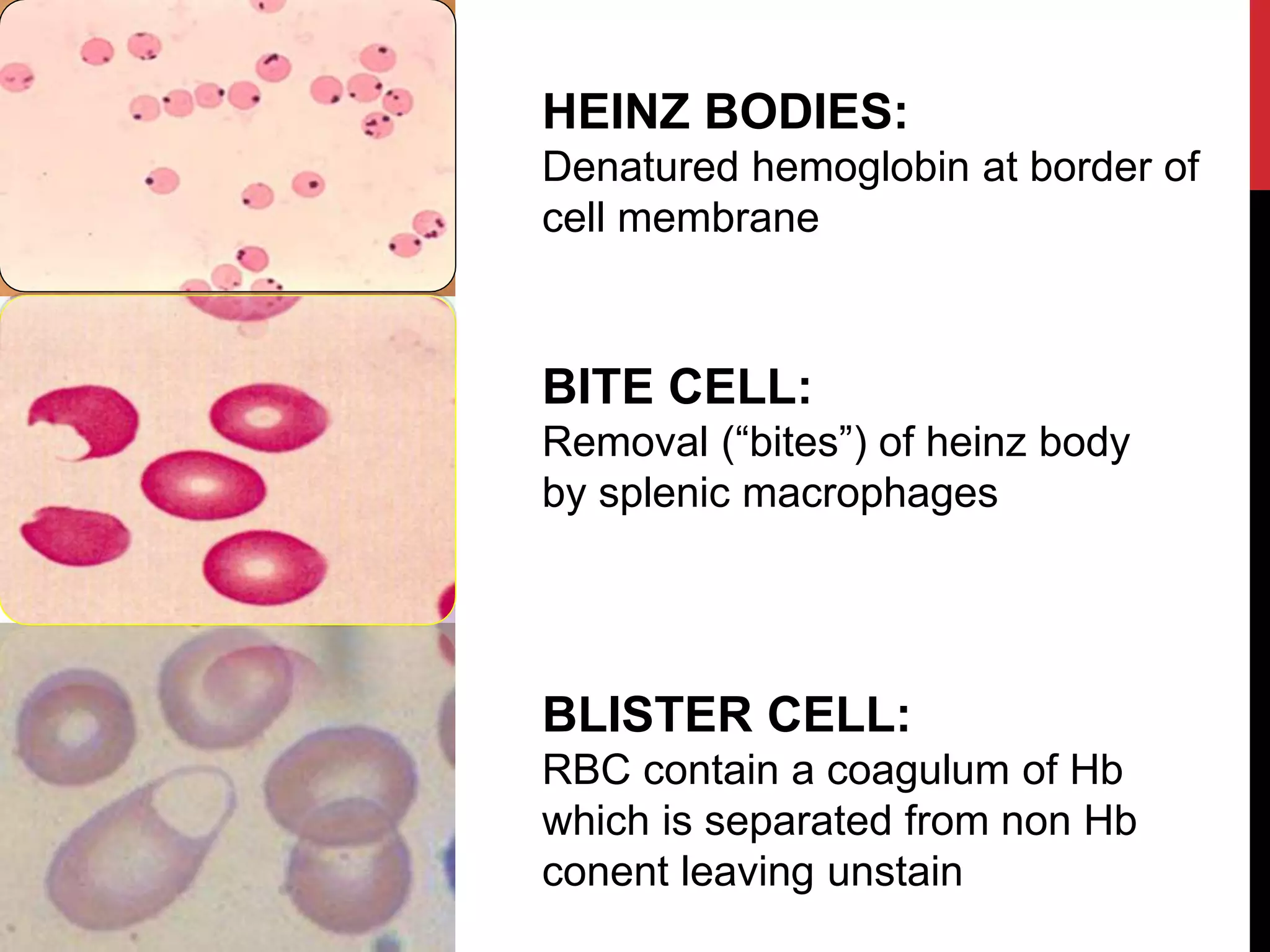
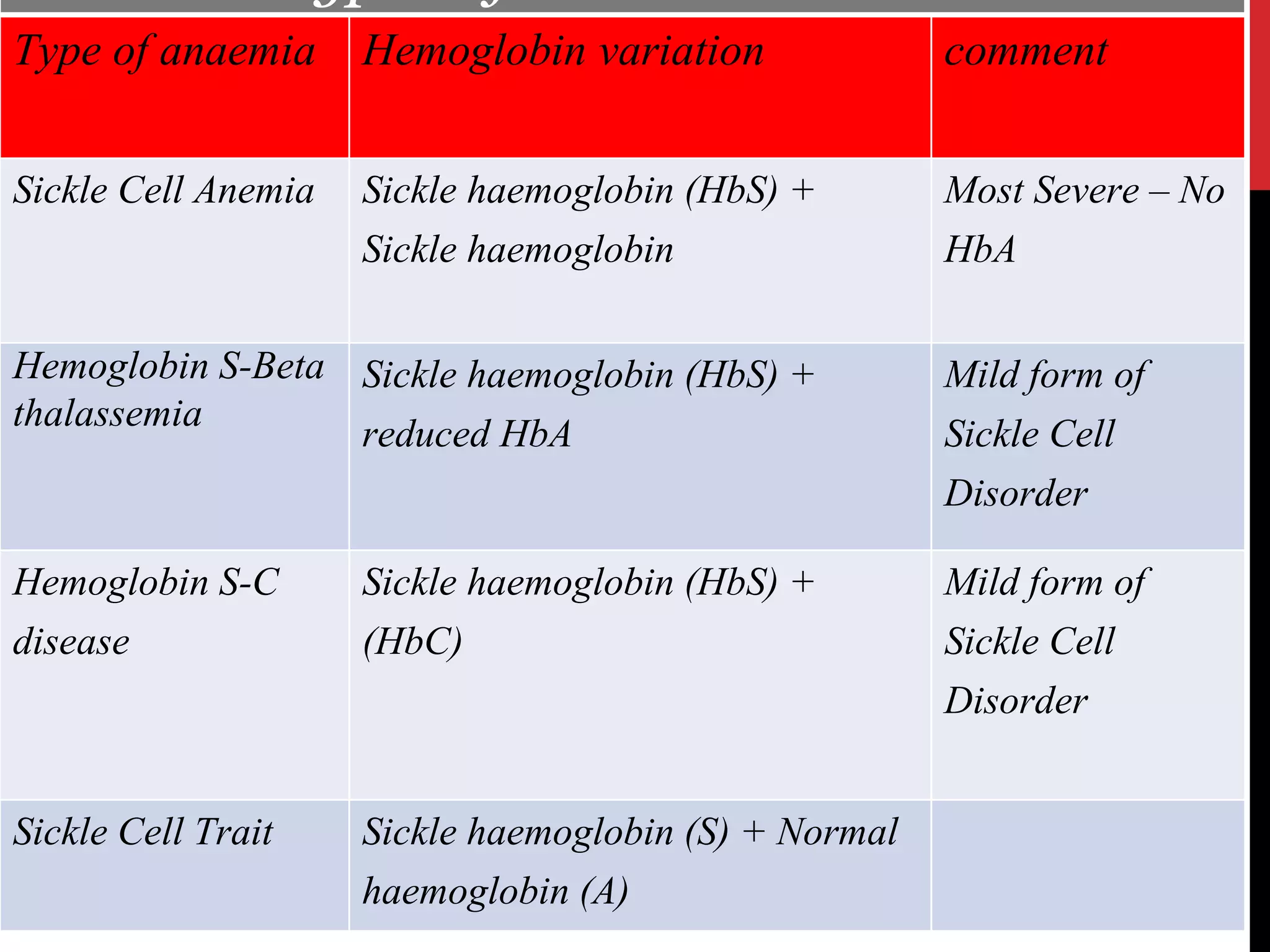
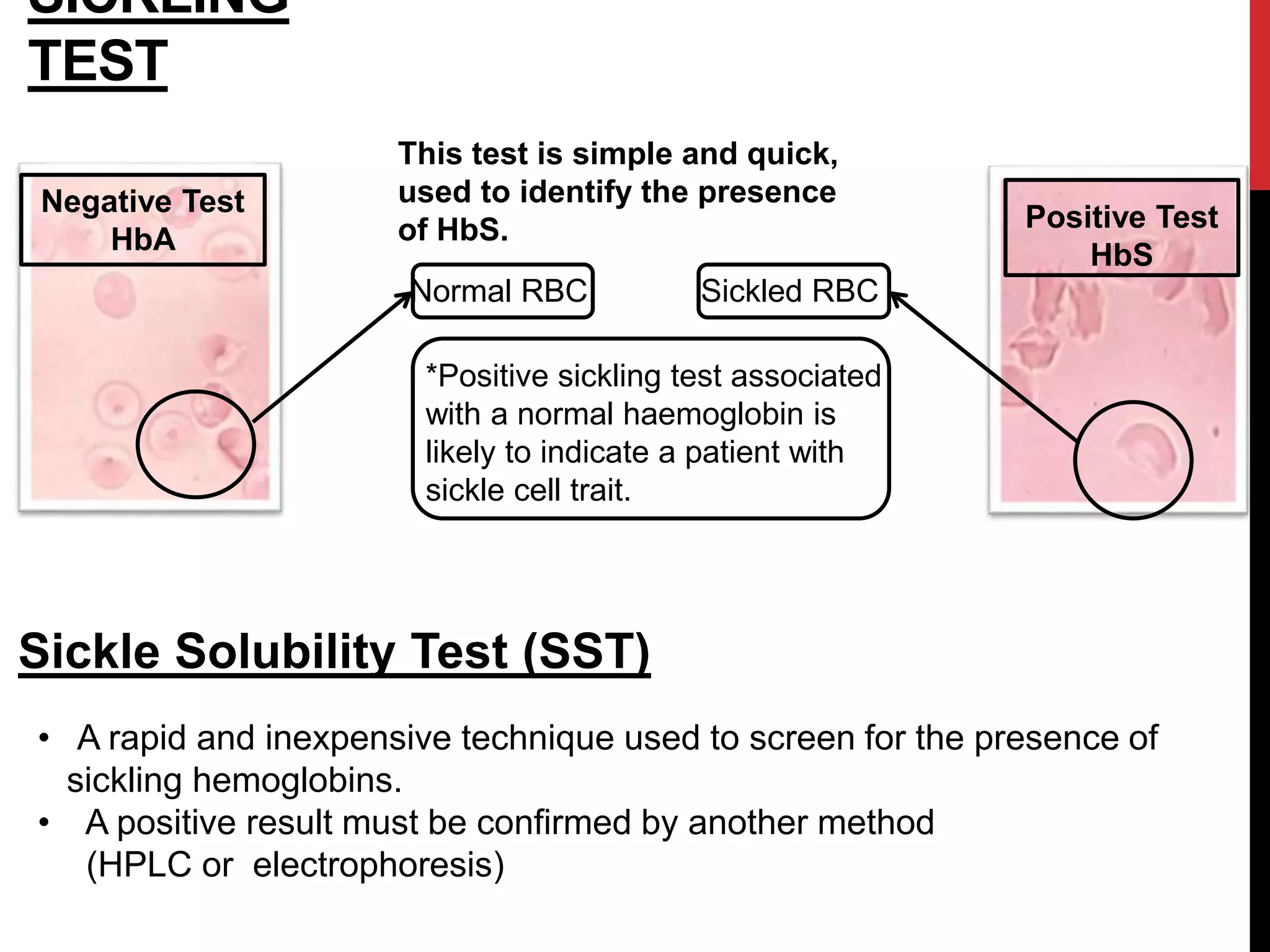
Hereditary hemolytic anemia can develop due to abnormal breakdown of red blood cells either in the blood vessels (intravascular hemolysis) or elsewhere in the body (extravascular). This document discusses the different types of hereditary hemolytic anemias including membrane-cytoskeletal defects like hereditary spherocytosis, enzymopathies like glucose-6-phosphate dehydrogenase deficiency, and hemoglobin defects like sickle cell disease. The causes, pathophysiology, clinical manifestations, and diagnostic testing for each condition are summarized.