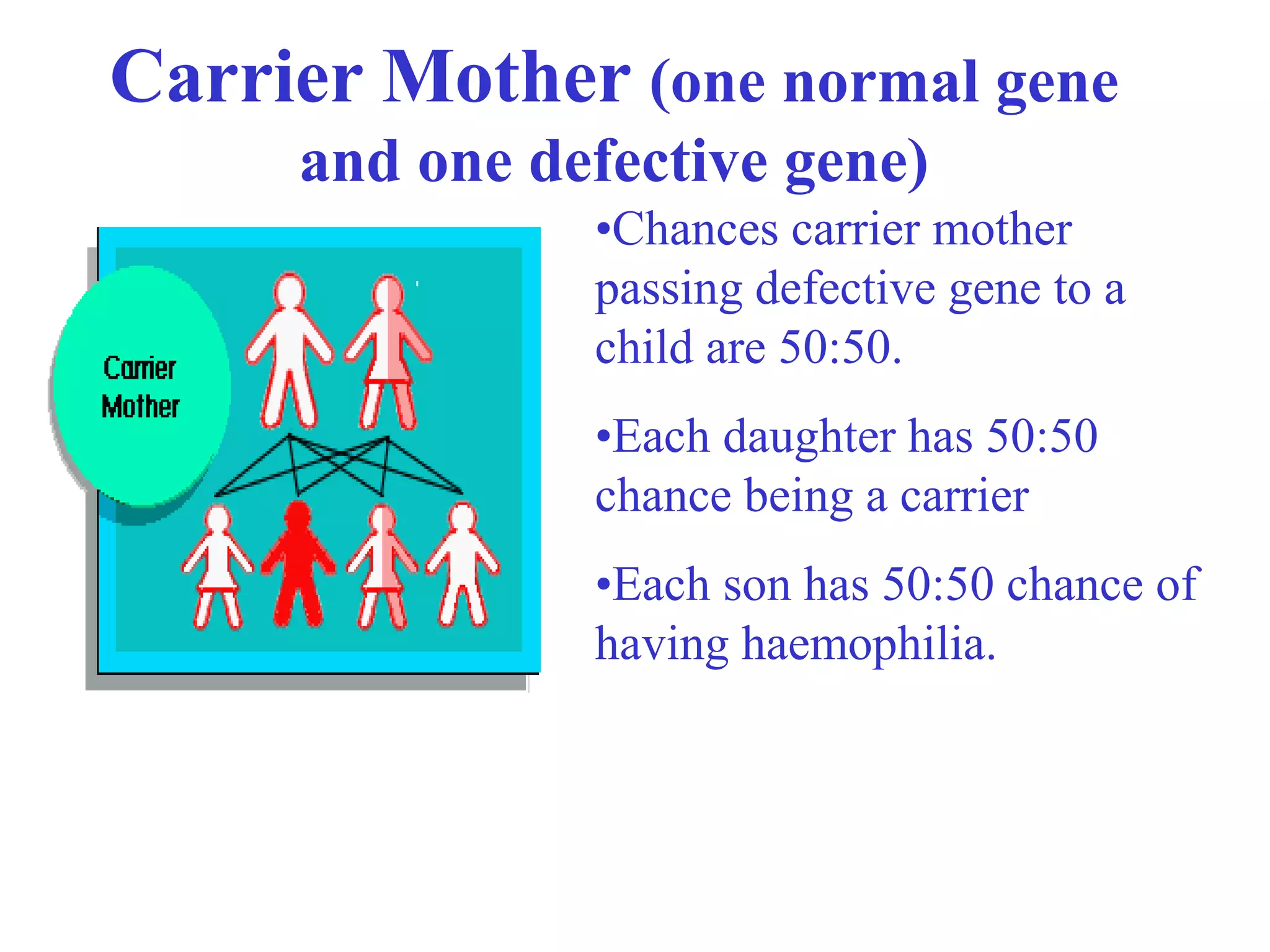
This document provides an overview of haemophilia and related bleeding disorders. It defines haemophilia as inherited blood disorders caused by a deficiency in specific clotting factors, either factor VIII (haemophilia A) or factor IX (haemophilia B). The severity of haemophilia depends on the level of deficient clotting factor, ranging from mild to severe. Treatment has advanced from no treatment to current use of recombinant clotting factors. Related disorders discussed include von Willebrand disease and platelet function defects.