Downloaded 508 times





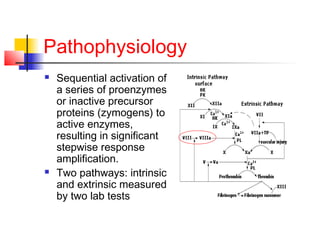
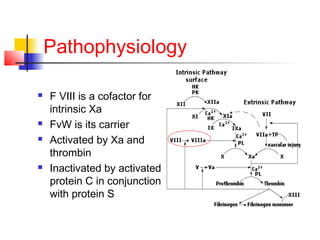
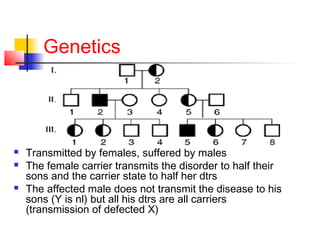





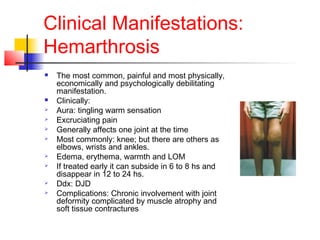


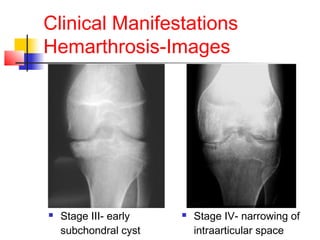


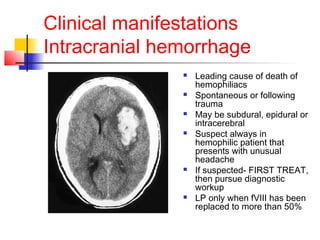

























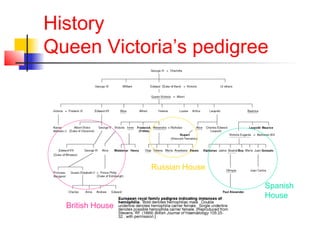




Hemophilia is a bleeding disorder caused by deficiencies in clotting factors VIII (hemophilia A) or IX (hemophilia B). The key manifestations are hemarthrosis, hematomas, and intracranial bleeding. It is inherited in an X-linked recessive pattern and treatment involves replacing the missing clotting factor through plasma-derived or recombinant sources. While gene therapy holds promise for a potential cure, current treatment focuses on factor replacement to prevent or treat bleeding episodes.









































































