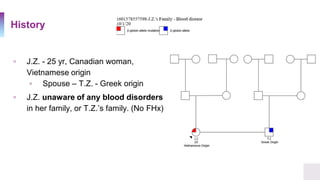
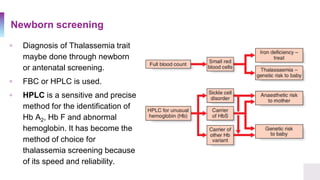
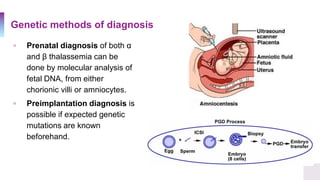
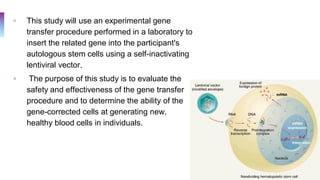
A 25-year-old Canadian woman of Vietnamese origin presented with mild microcytic anemia during routine prenatal care. Testing revealed she had beta-thalassemia trait due to a nonsense mutation in her beta-globin gene. Her Greek husband also carried the same mutation, giving them a 25% risk of having a child with beta-thalassemia major. Beta-thalassemia is caused by a deficiency in alpha or beta globin production and results in anemia and other symptoms. Current treatments include blood transfusions, chelation therapy, and potentially gene therapy through stem cell gene correction.
![The Case
▫ A 25 year old Healthy Canadian woman, presented to her
obstetrician for routine prenatal care.
▫ Results of her complete blood count showed signs of a mild
microcytic anemia
▫ Hemoglobin 98 g/L [ref. 121-151 g/L]
▫ Mean Corpuscular Volume 75 µm³ [ref. 87 ± 5]](https://image.slidesharecdn.com/thalassemia-dilinaaarewattecase44-201008082325/85/Thalassemia-Case-Presentation-2-320.jpg)




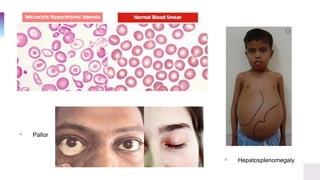
![Clinical Symptoms & Diagnosis
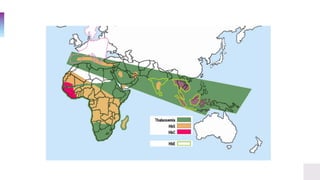
▫ In a population, the α-globin mutations are reflected by the
phenotypes observed in the population.
▫ Clinical symptoms
▫ Anemia [hypochromic
microcytic]
▫ Pallor
▫ Fatigue
▫ Hepatosplenomegaly](https://image.slidesharecdn.com/thalassemia-dilinaaarewattecase44-201008082325/85/Thalassemia-Case-Presentation-9-320.jpg)







































![THALASEMIA definition and pathophysiologyd].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/thalasemia-240515044127-8ce804c5-thumbnail.jpg?width=640&height=640&fit=bounds)






















![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)
![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)











![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)





