Downloaded 1,002 times





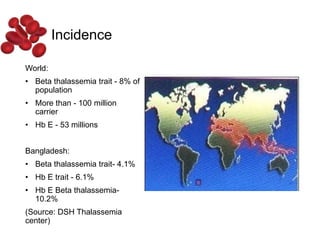
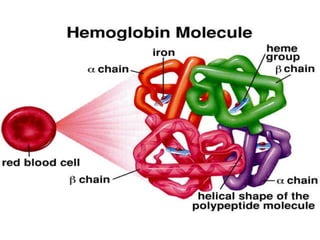
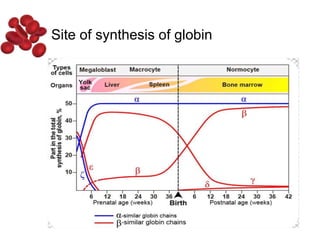
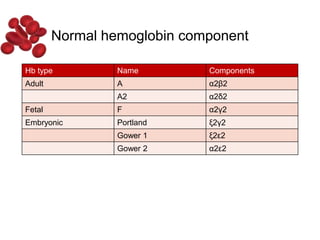
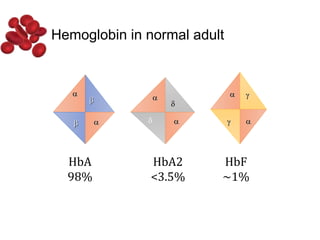
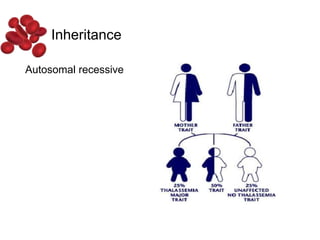
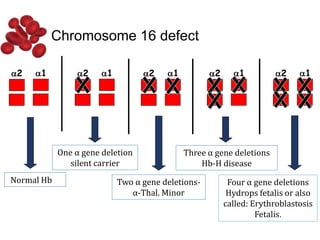
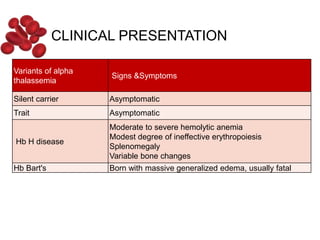

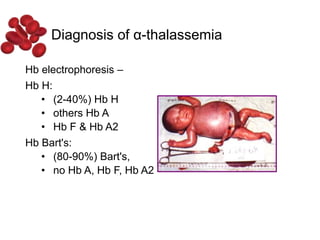

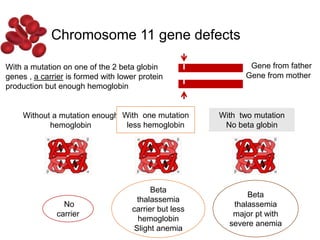

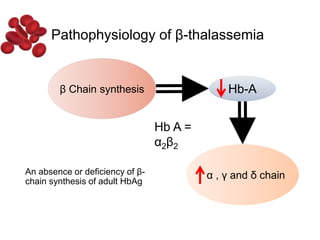
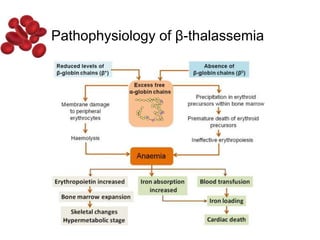
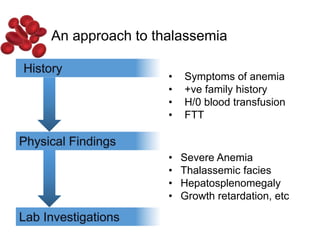
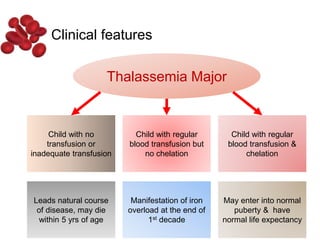
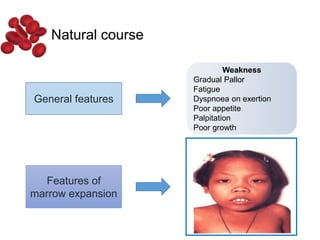
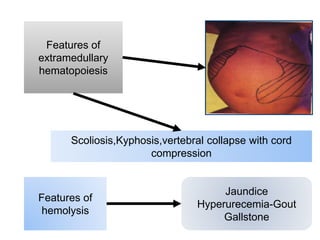
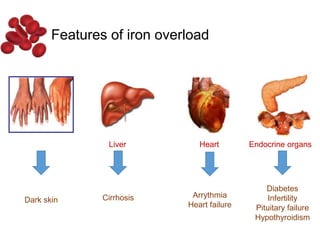

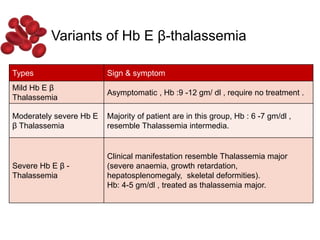

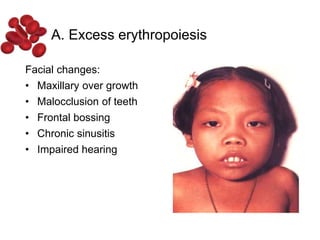
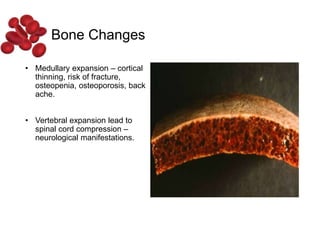
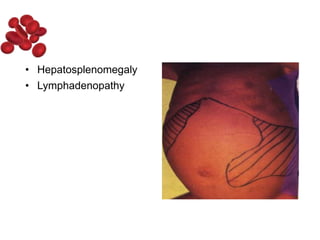
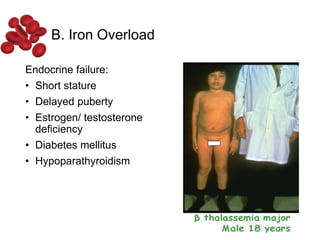
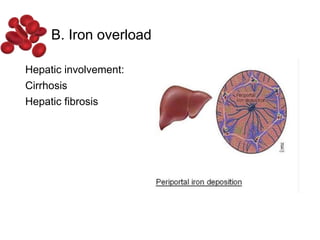




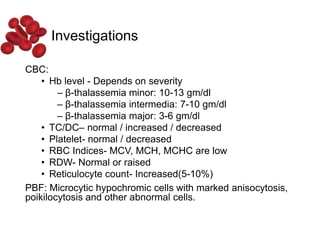
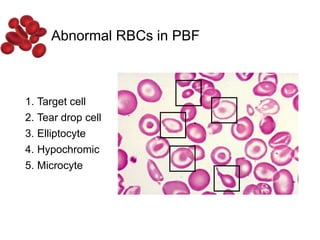
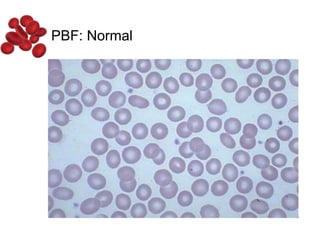
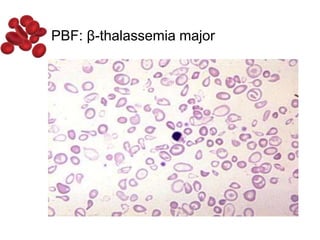
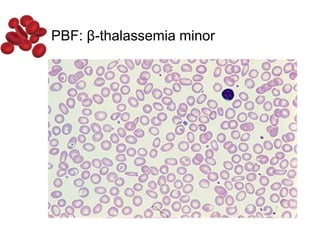

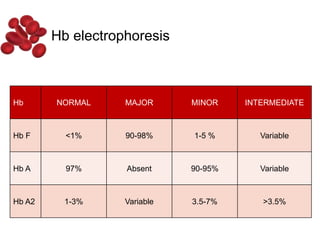
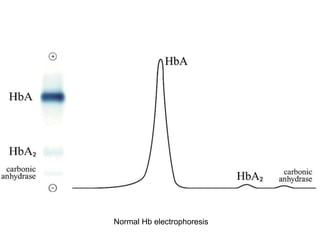
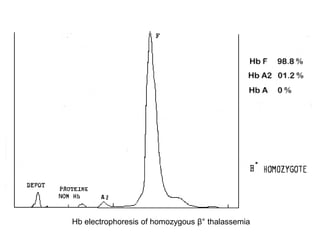
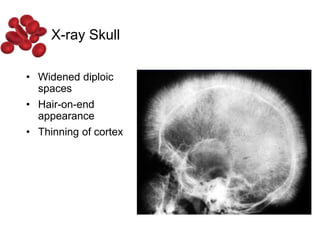
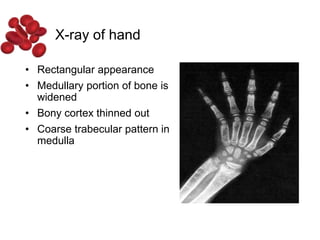

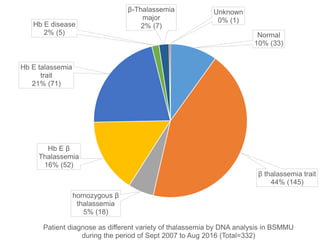
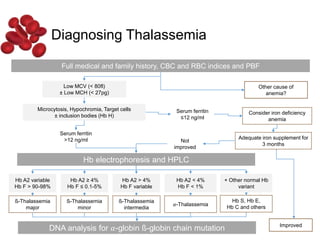
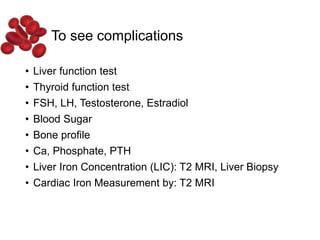
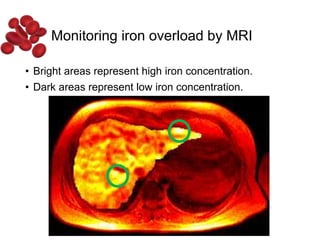


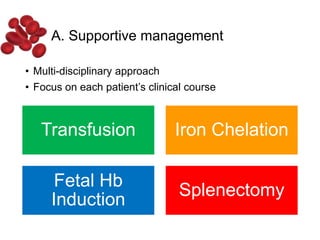







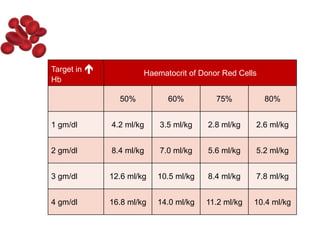
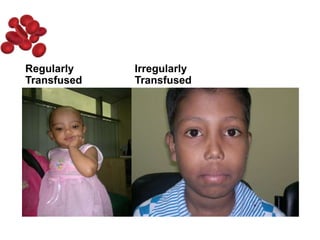




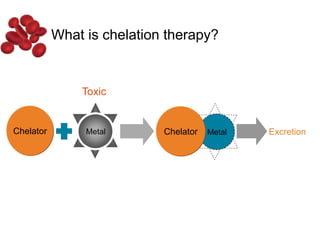
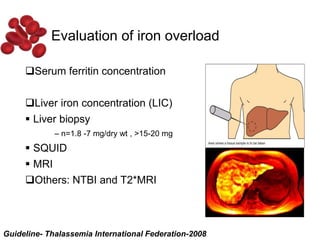


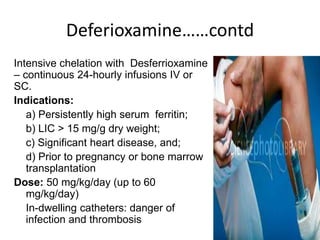






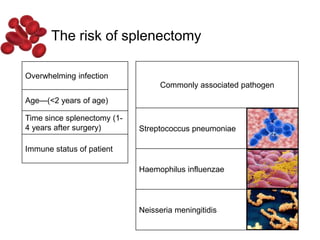

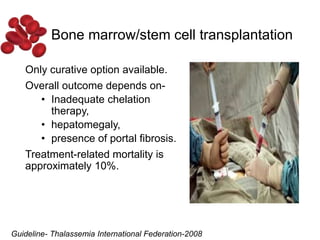

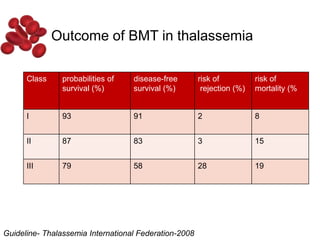


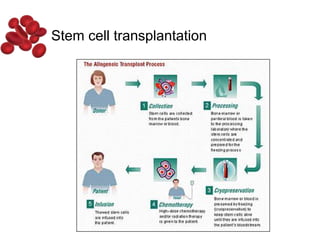











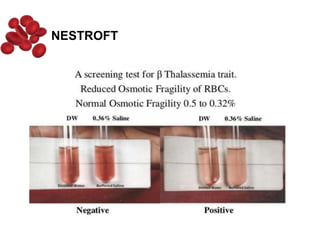


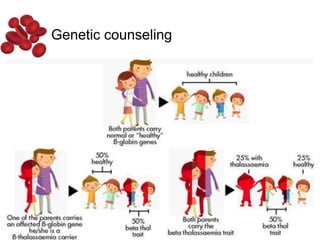
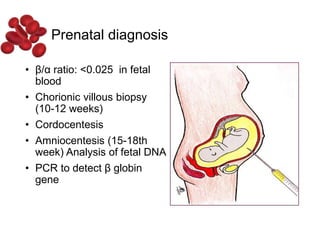
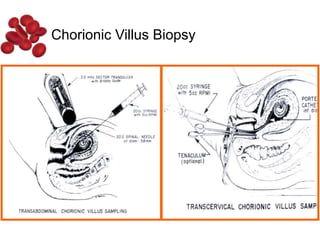






Halima, an 11-year-old girl with consanguineous parents, presented with not growing well, gradual pallor, and abdominal distension for 7 years. On examination, she was severely pale with facial dysmorphism and hepatosplenomegaly. Her history included repeated blood transfusions. She was diagnosed with hereditary hemolytic anemia. The seminar discussed thalassemia, including the types of thalassemia, clinical features, investigations, complications, and management with a focus on blood transfusions and chelation therapy.




































































































