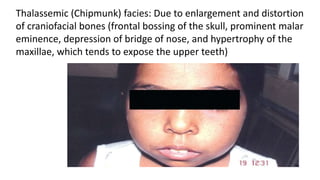
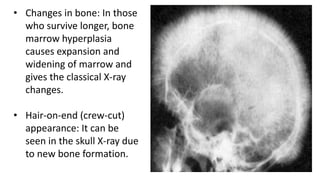
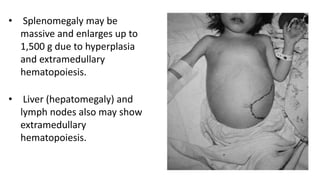
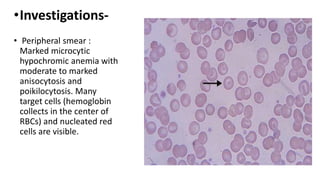
Thalassemia is a group of inherited blood disorders caused by reduced or absent globin chain synthesis. It is classified into alpha or beta thalassemia depending on the defective chain. Alpha thalassemia results from deletion of one or more of the four alpha globin genes and ranges from silent carrier to fatal hydrops fetalis. Beta thalassemia major is due to absent or reduced beta chain synthesis and causes severe anemia requiring lifelong blood transfusions and iron chelation therapy to prevent organ damage from iron overload. Complications include bone abnormalities, extramedullary hematopoiesis, and failure to thrive if untreated.