Download as PDF, PPTX



































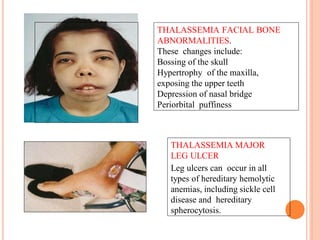





























There are two main types of thalassemia - alpha and beta thalassemia. Alpha thalassemia is caused by a deficiency or absence of alpha globin chains, while beta thalassemia results from a defective or reduced production of beta globin chains. Thalassemia is inherited in an autosomal recessive pattern and is common in Mediterranean, Middle Eastern, Asian and African populations. Regular blood transfusions along with iron chelation therapy are the mainstay of treatment for thalassemia major to prevent complications from anemia and iron overload. Complications can affect the heart, liver, endocrine system and bones if not properly managed.














































































