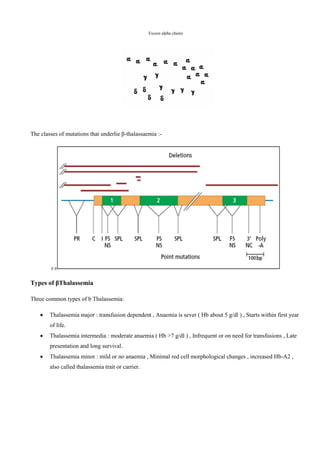
The document primarily discusses thalassemia, an inherited blood disorder caused by impaired synthesis of globin chains, leading to anemia and affecting individuals of Mediterranean, Middle Eastern, African, and Southeast Asian descent. It details the types of thalassemia, specifically alpha and beta thalassemia, the genetic mechanisms behind them, their incidence in populations, symptoms, diagnosis, treatment options, and prevention strategies. The conclusion emphasizes thalassemia as a common genetic disorder, highlighting the need for awareness and management strategies.