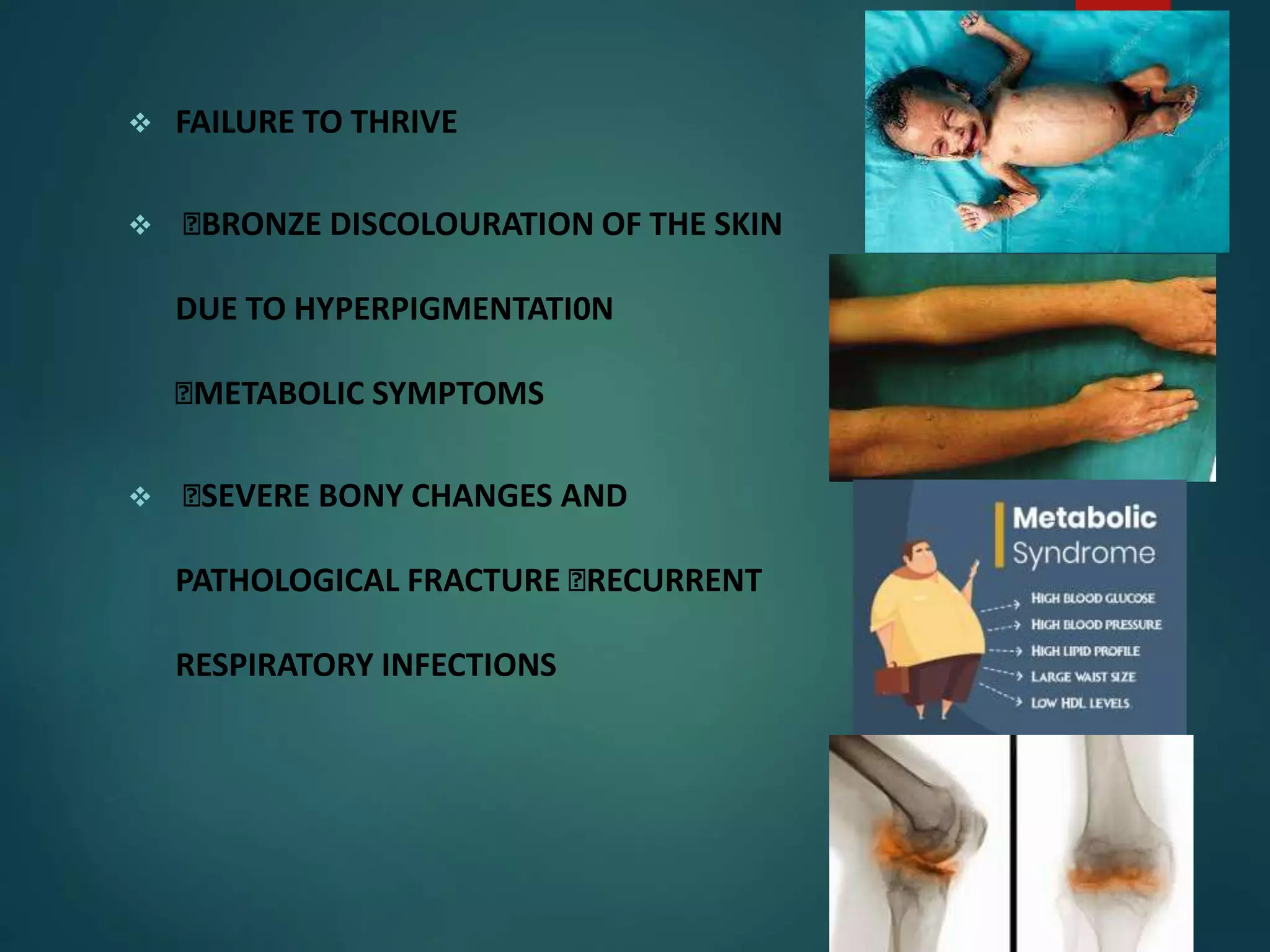
Thalassemia is a genetic blood disorder characterized by insufficient hemoglobin production, leading to severe anemia and organ oxygen deprivation. The two main types, alpha and beta thalassemia, result from genetic alterations affecting the alpha and beta globin chains, respectively, with varying severity from mild symptoms to life-threatening conditions. Management includes regular blood transfusions, iron chelation therapy, and potential surgical interventions, while preventive measures focus on genetic counseling and awareness to improve life expectancy and quality of life for affected individuals.







































































![THALASEMIA definition and pathophysiologyd].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/thalasemia-240515044127-8ce804c5-thumbnail.jpg?width=640&height=640&fit=bounds)




































