
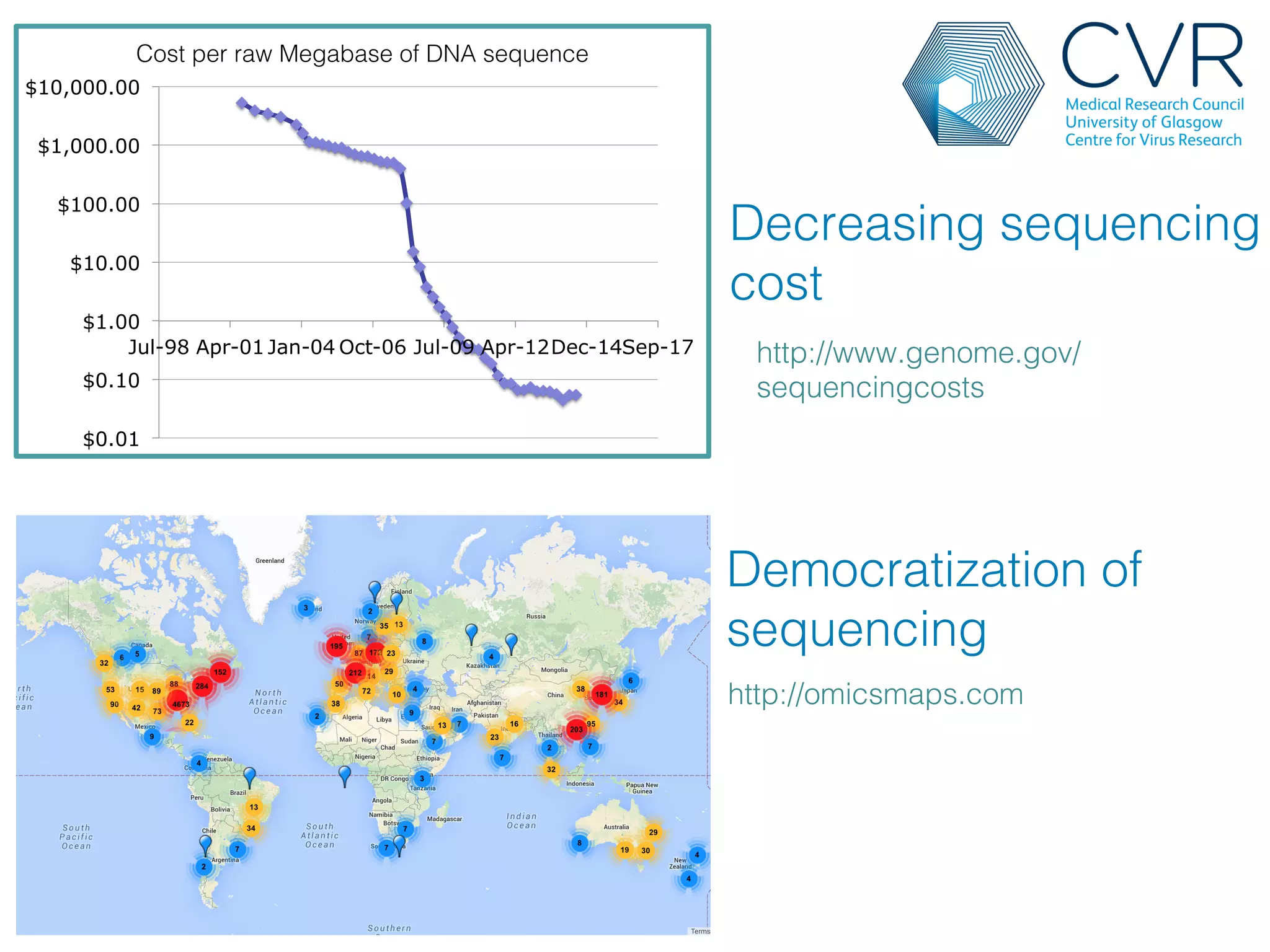


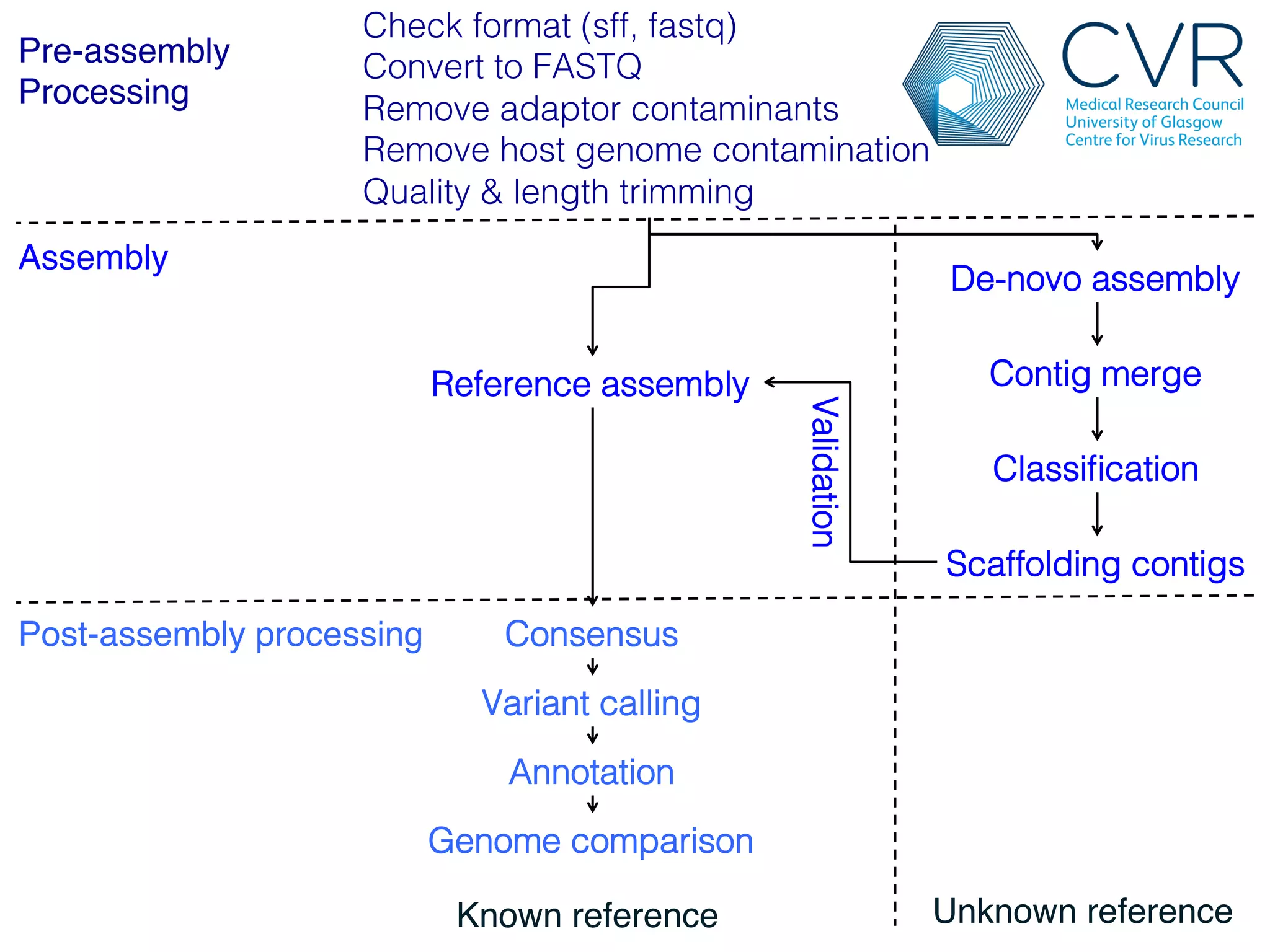

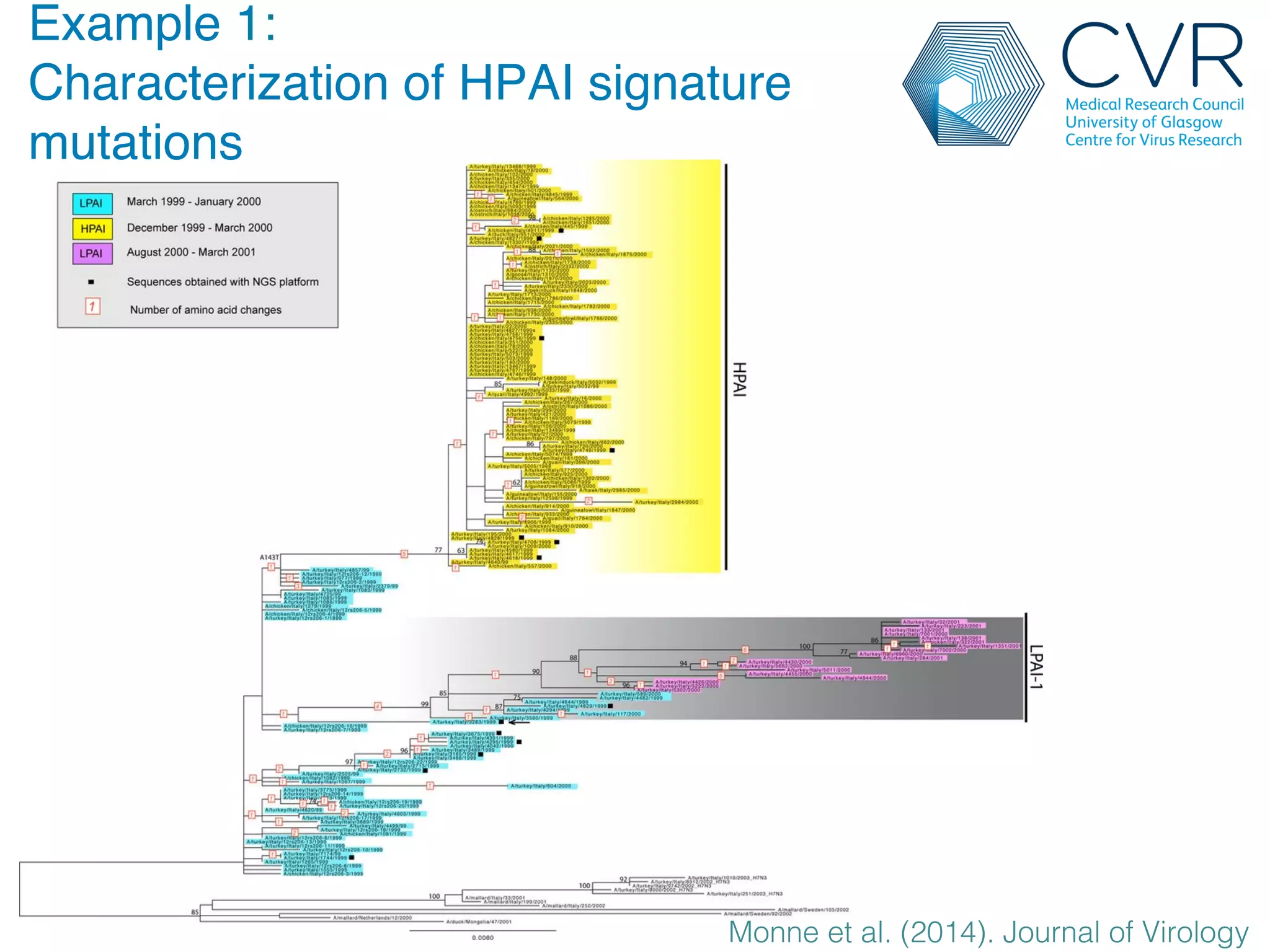
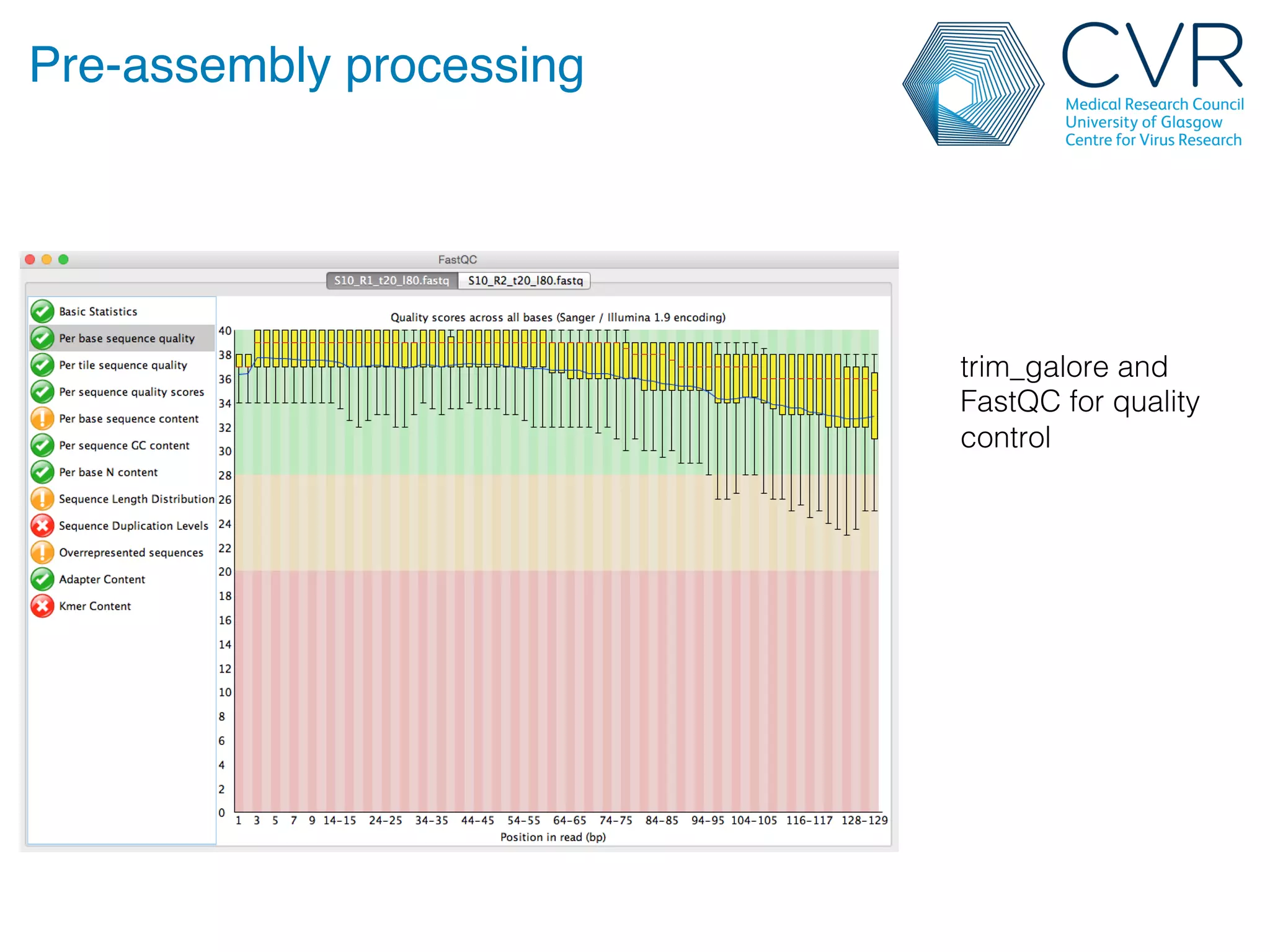

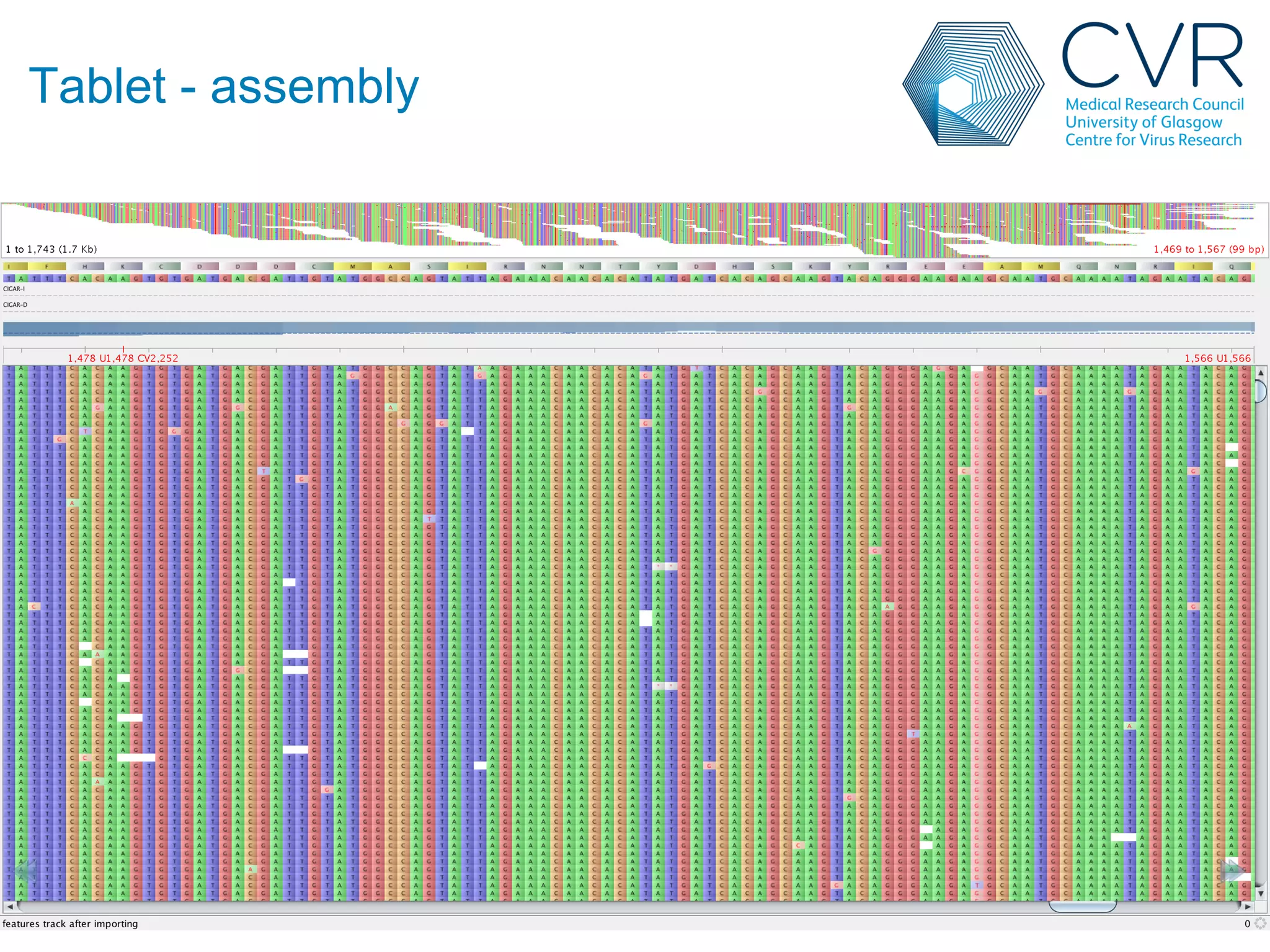
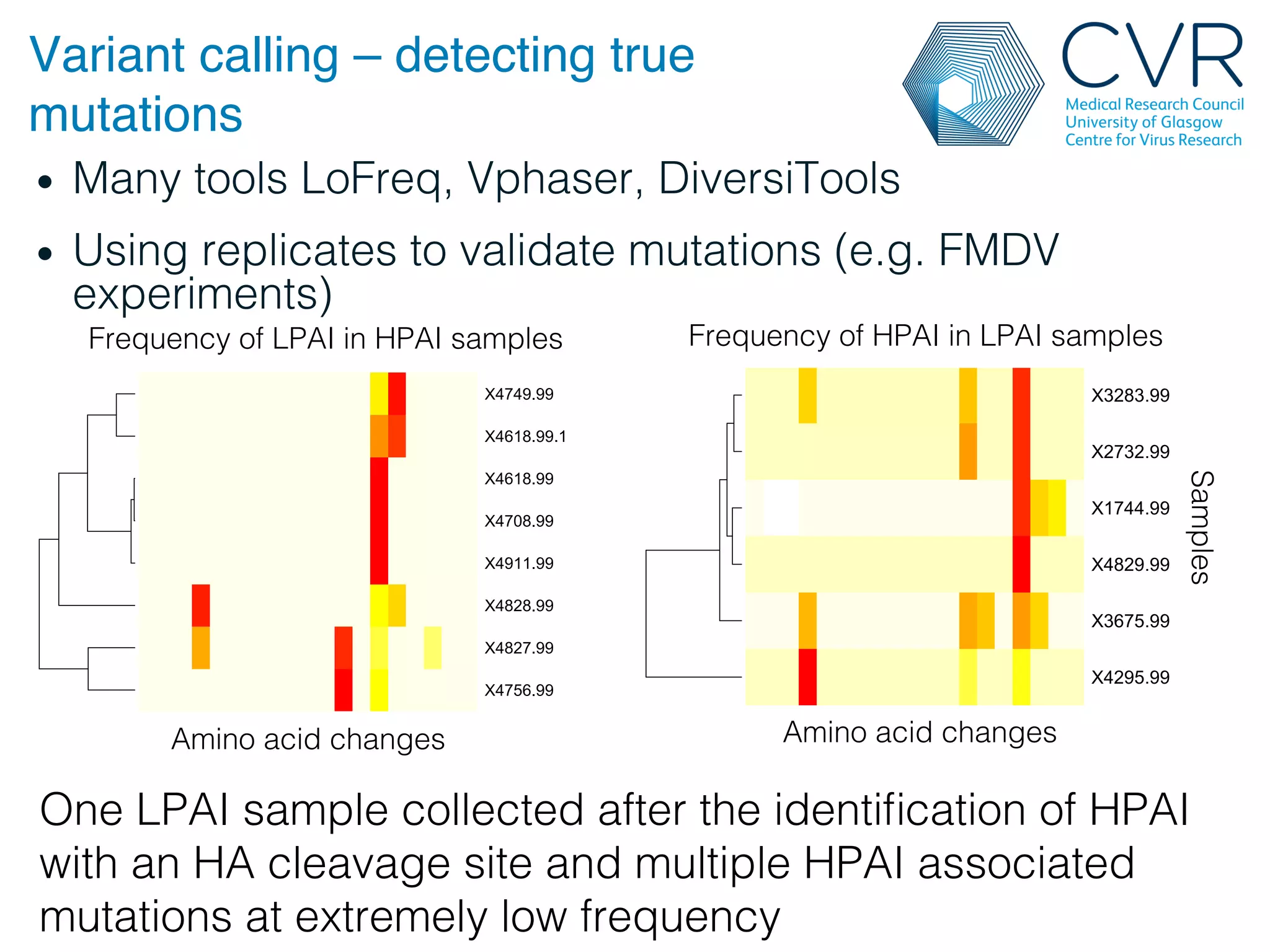


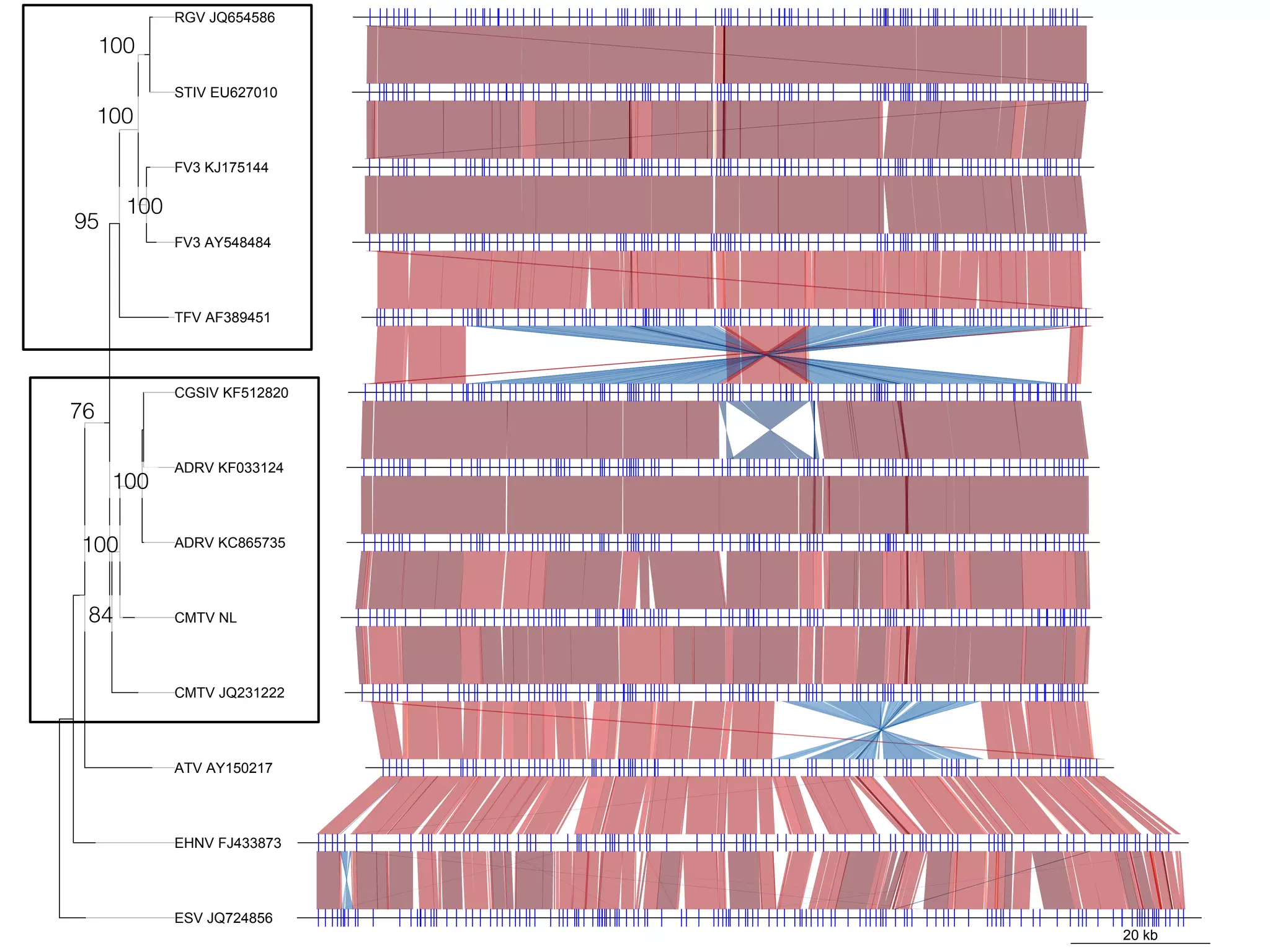

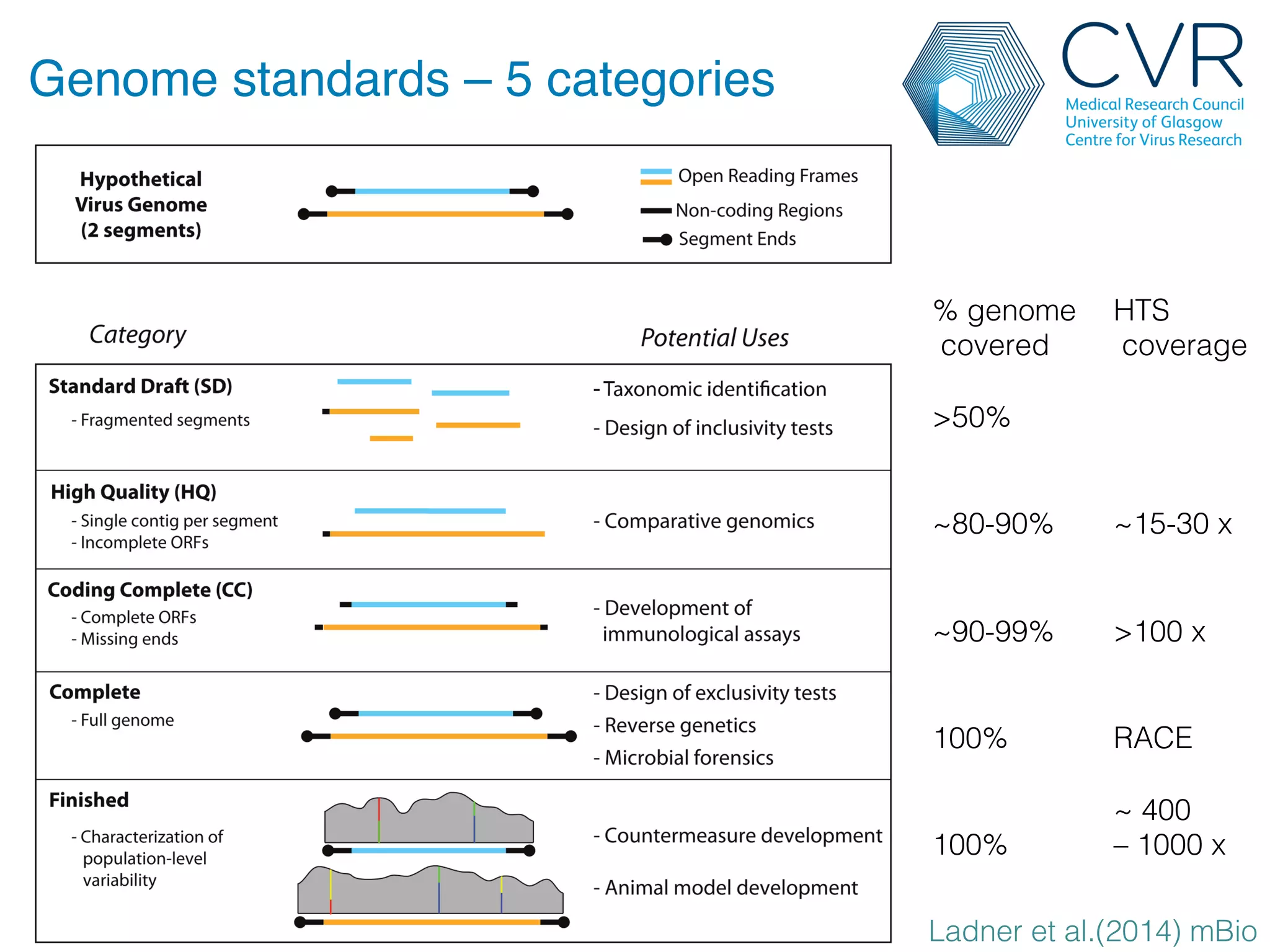
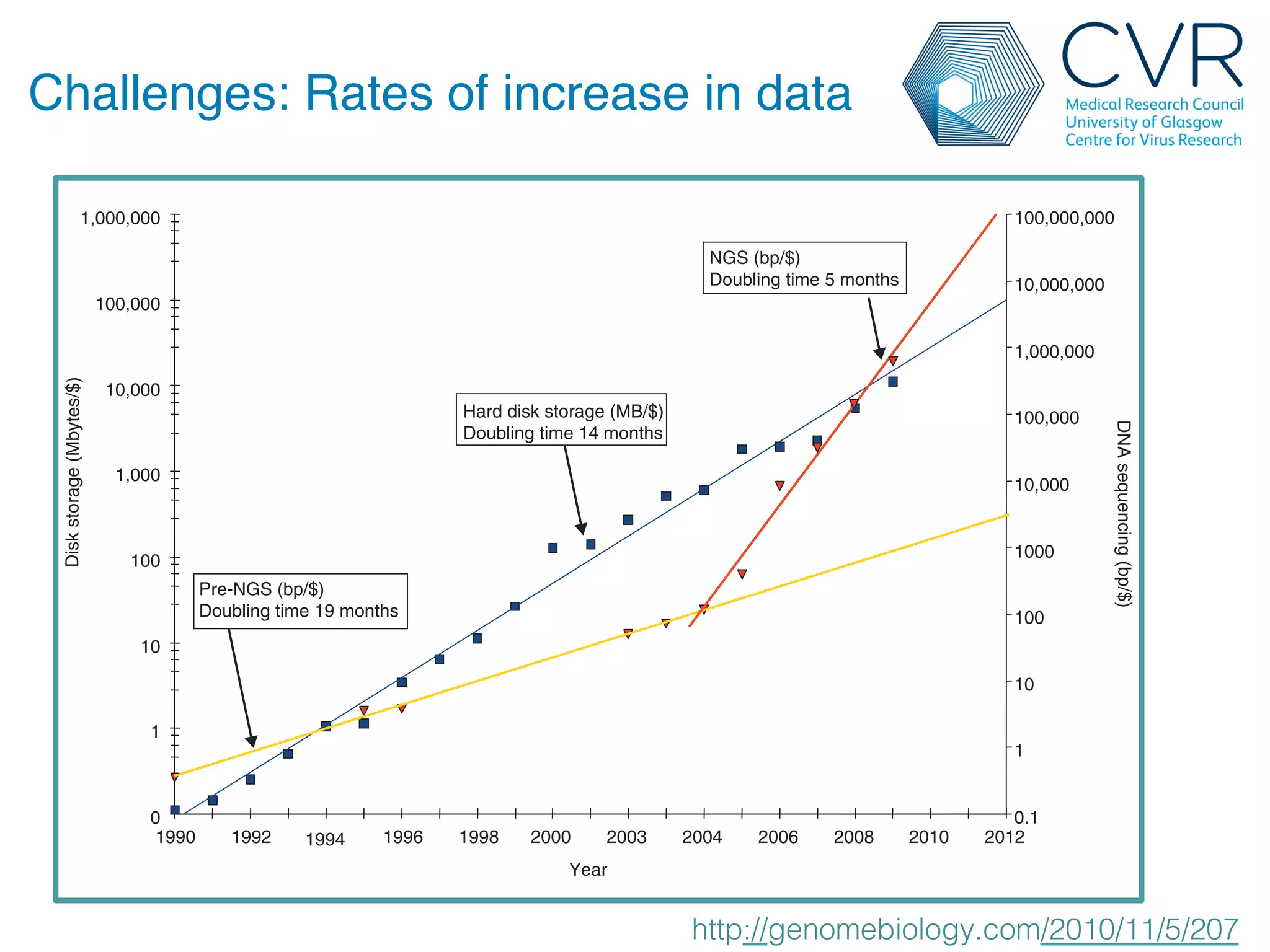



The document discusses the technological advancements in DNA sequencing, including decreasing costs and the democratization of sequencing methods. It outlines various applications of high-throughput sequencing, such as whole genome sequencing and metagenomics, while emphasizing the importance of flexible and accurate genome assembly pipelines. Additionally, it highlights challenges in data management and the necessity for dedicated bioinformaticians in laboratories to handle increased data complexity.



































































