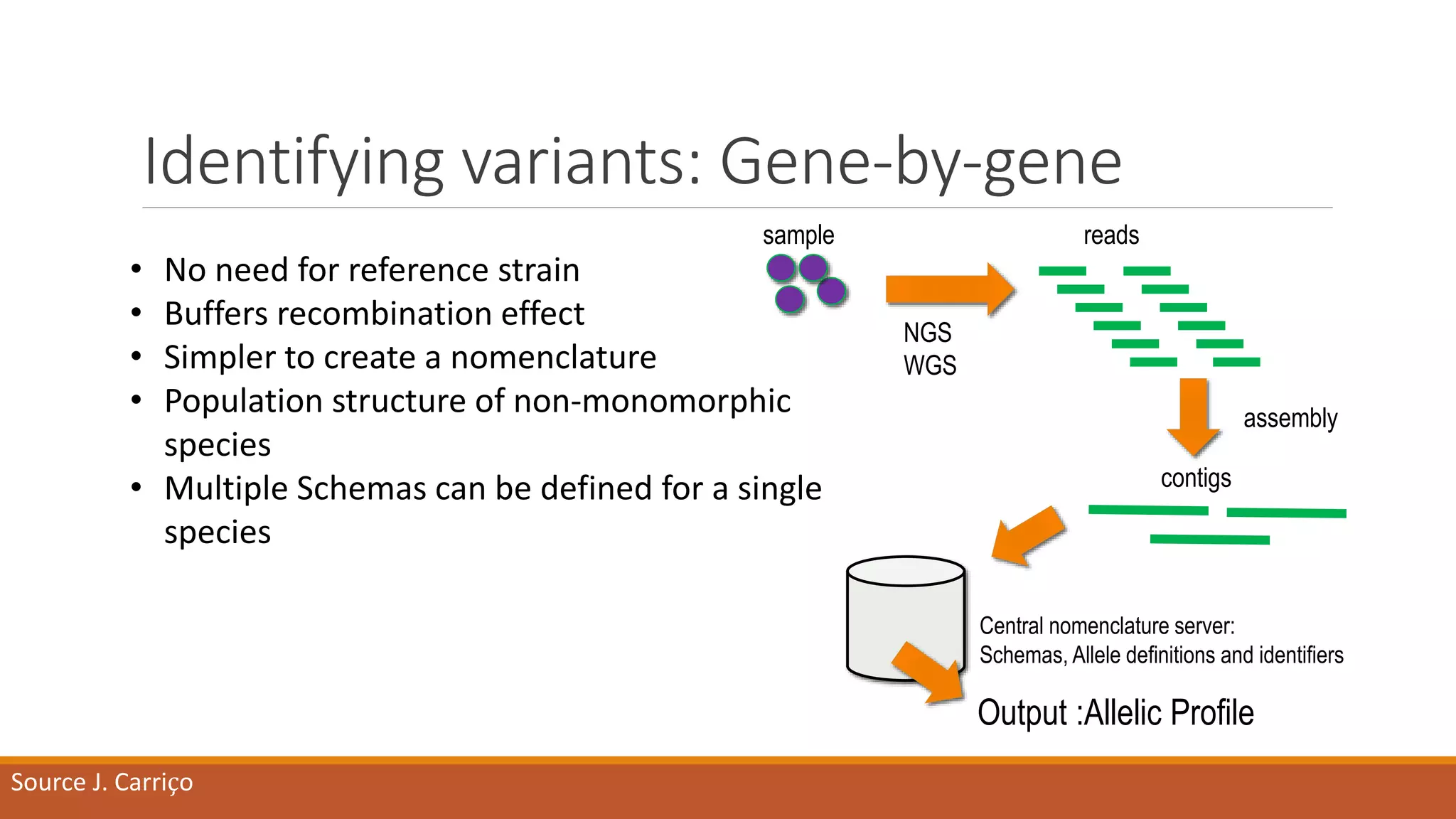
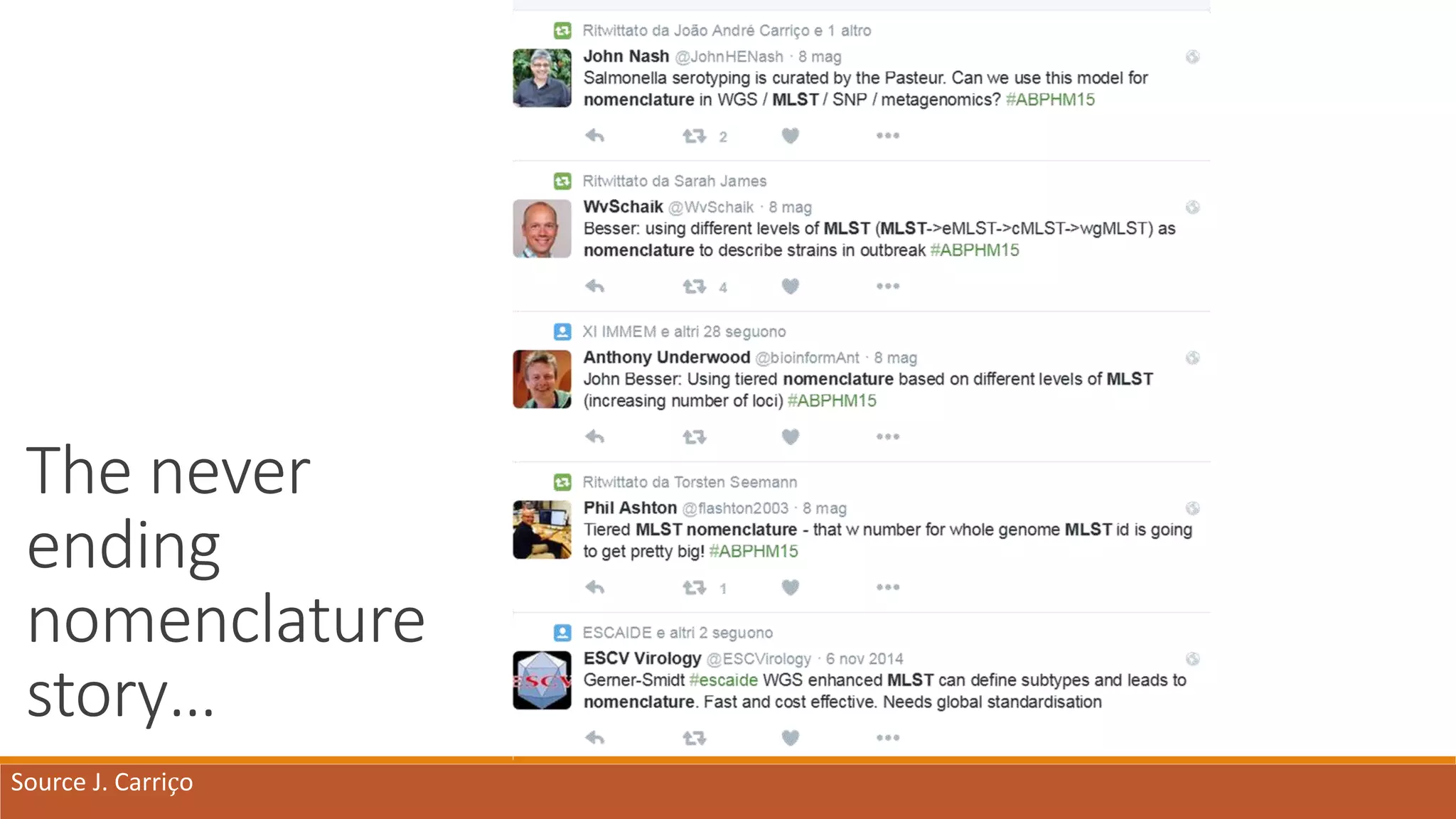
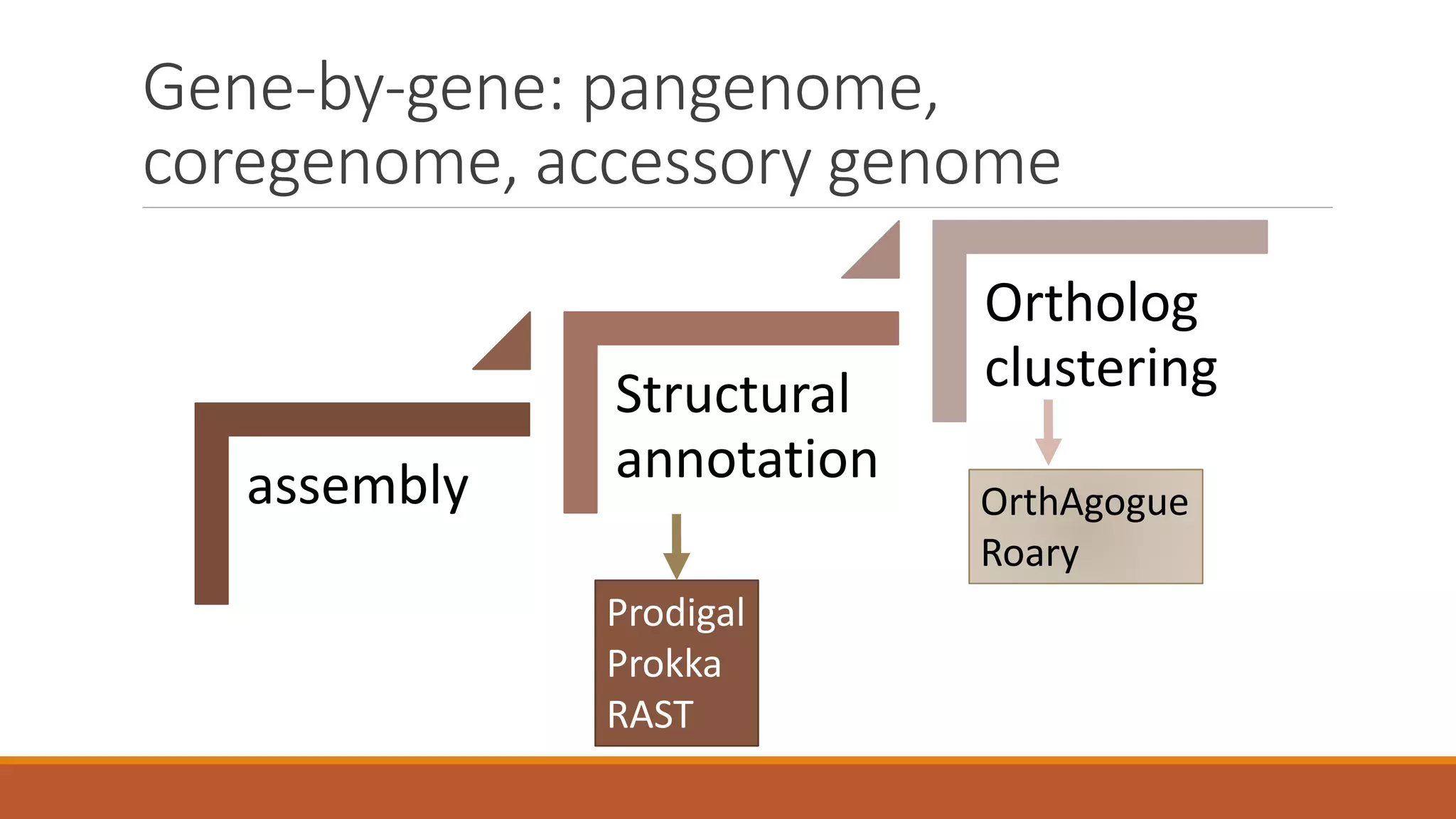
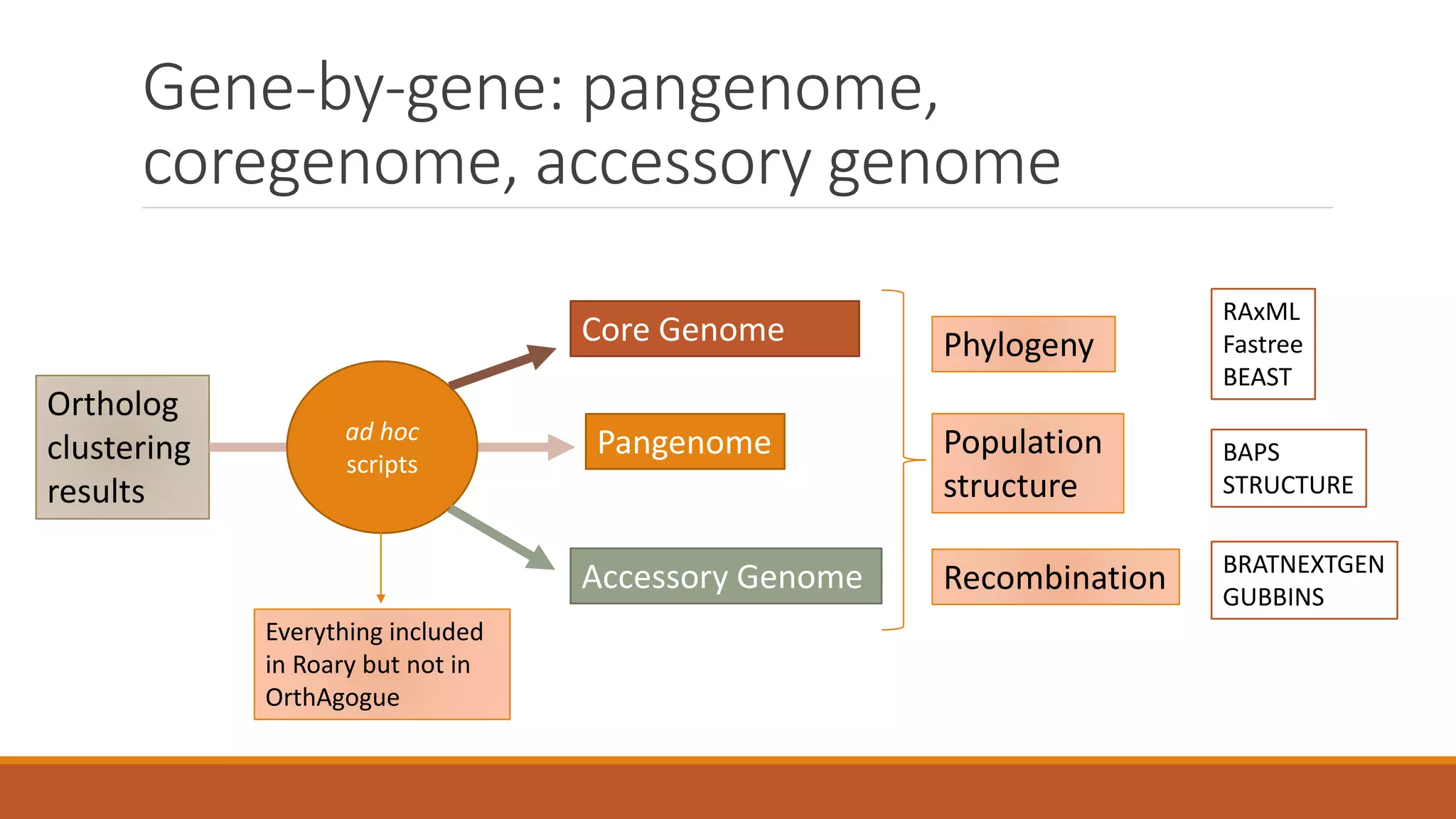
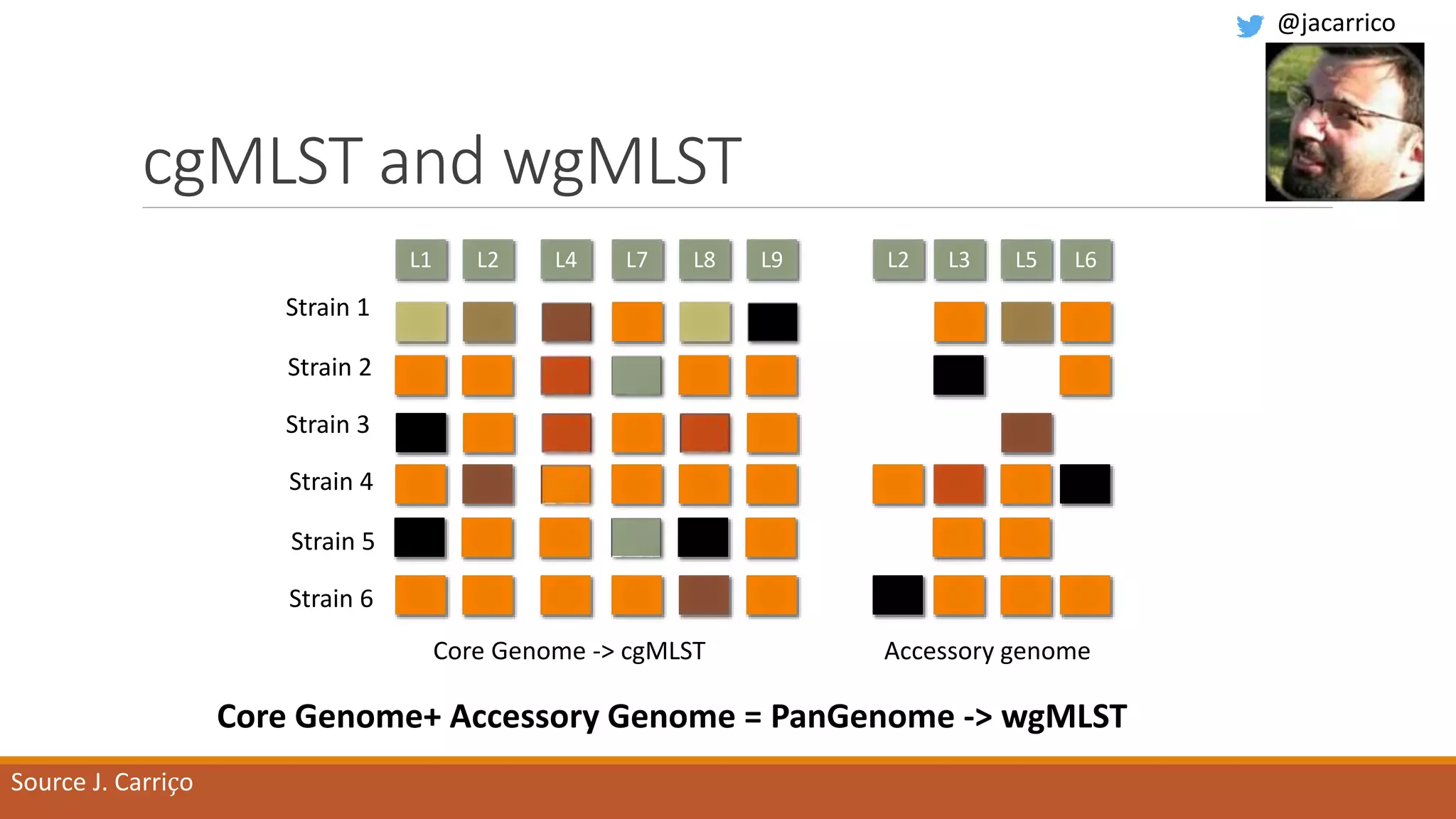
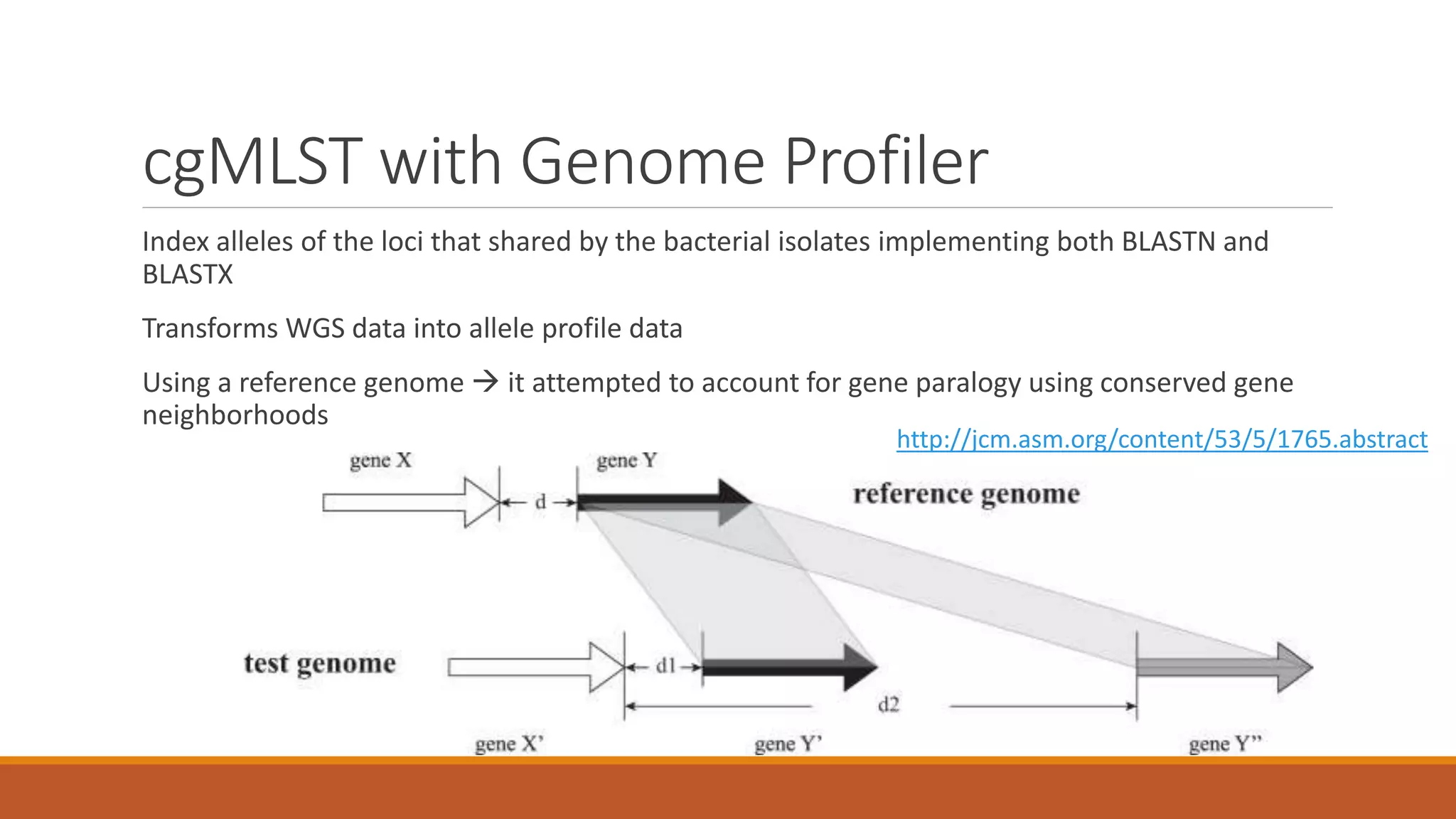
This document provides an overview of tools and approaches for analyzing bacterial population genomics and evolution using next-generation sequencing (NGS) data. It discusses identifying variants from NGS reads using SNP-based or gene-by-gene approaches. It also covers assembly-free and assembly-based analyses, including tools for short-read assembly, pangenome alignment, core genome alignment, and ortholog clustering. Population genomics applications like cgMLST/wgMLST, population structure analysis, and recombination detection are also briefly introduced. The document aims to provide bacterial genomics researchers with a toolbox of software and strategies for population analysis using NGS data.