
Recommended
More Related Content
What's hot
What's hot (20)
Similar to Sequence assembly
Similar to Sequence assembly (20)
Recently uploaded
Recently uploaded (20)
Sequence assembly
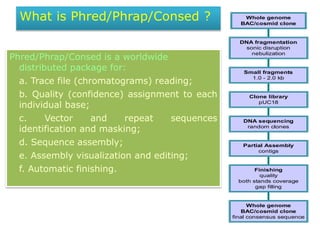
- 1. What is Phred/Phrap/Consed ? Phred/Phrap/Consed is a worldwide distributed package for: a. Trace file (chromatograms) reading; b. Quality (confidence) assignment to each individual base; c. Vector and repeat sequences identification and masking; d. Sequence assembly; e. Assembly visualization and editing; f. Automatic finishing. Whole genome BAC/cosmid clone final consensus sequence Finishing quality both stands coverage gap filling Partial Assembly contigs DNA sequencing random clones Clone library pUC18 Small fragments 1.0 - 2.0 kb DNA fragmentation sonic disruption nebulization Whole genome BAC/cosmid clone
- 3. Trace File High quality region – no ambiguities (Ns) - no ambiguities (Ns) - no noise - peaks very well spaced
- 4. Trace File Medium quality region – some ambiguities (Ns)
- 5. Trace File Poor quality region – low confidence - some ambiguities (Ns) - bad noise (notice baseline) - overlapping peaks - can be caused by bad quality template, bad matrix, low signal to noise rate
- 6. Trace File Poor quality region – low confidence Poor quality read: - many ambiguities (Ns) - noise - caused by homopolymeric region/polymerase slippage
- 7. Sudden drop artifact: - good quality region is followed by a sudden drop of signal - caused by secondary structure
- 8. Sequence Assembly The phred software reads DNA sequencing trace files, calls bases, and assigns a quality value to each base. The quality value is a log-transformed error probability, specifically Q = -10 log10( Pe ) where Q and Pe are respectively the quality value and error probability of a particular base call. Phred can use the quality values to perform sequence trimming. Phred works well with trace files from the most manufacturers' sequencing machines The program was developed by Drs. Phil Green and Brent Ewing, and is copyrighted by the University of Washington
- 9. Phred is generates highly accurate, base-specific quality scores Quality scores range from 4 to about 60, with higher values corresponding to higher quality Phred quality score Probability that the base is called wrong Accuracy of the base call 10 1 in 10 90% 20 1 in 100 99% 30 1 in 1,000 99.9% 40 1 in 10,000 99.99% 50 1 in 100,000 99.999% Ideal tool to assess the quality of sequences The most commonly used method is to count the bases with a quality score of 20 and above (sometimes called "high quality bases"); the resulting number is often called the "Phred20 score"
- 10. Conversion of phd files into FASTA files phd2fasta script Features: - Phred creates single-sequences files containing the sequence itself plus the quality assignments (phd files) - The input file for cross_match and phrap programs is a multiple sequence file in FASTA format - A Perl script named phd2fasta converts the phd files into two multiple sequence FASTA format files, containing the sequence information and the basecall quality information respectively - phredPhrap script automatically executes phd2fasta before running cross_match and phrap!
- 11. Phrap Phragment Assembly Program or… Phil’s Revised Assembly Program Phrap is a program for assembling shotgun DNA sequence data Key Features: a. Uses the entire read content – no need for trimming. b. User supplied (i.e. Repbase) + internally computed data – better accuracy of assembly in the presence of repeats. c. Contig sequence is constituted by a mosaic of the highest quality parts of the reads – it’s not a consensus!
- 12. Phrap is a program for assembling shotgun DNA sequence data.. Accurate consensus sequences. Phrap uses Phred's quality scores to determine highly accurate consensus sequences. Phrap examines all individual sequences at a given position, and generally uses the highest quality sequence to build the consensus - similar to the way scientists would correct consensus sequences during "contig editing". Consensus quality estimates. Phrap uses the quality information of individual sequences to estimate the quality of the consensus sequence. In addition, Phrap uses available information about sequencing chemistry (dye terminator or dye primer) and confirmation by "other strand" reads in estimating the consensus quality. This often allows scientists to ignore random errors, and to focus finishing efforts exclusively onto regions where the data quality is insufficient. Consensus quality estimates can also be very helpful in mutation detection by DNA sequencingAbility to assemble very large projects. Phrap has been used routinely to assembly bacterial genomes sequenced by the "shotgun" approach, where each project contained tens of thousands of reads. Smaller bacterial genomes (2 million bases or less) could often be assembled in less than three hours.
- 13. Improved identification and handling of repeats. Phrap uses quality scores to estimate whether discrepancies between two overlapping sequences are more likely to arise from random errors, or from different copies of a repeated sequence. For repeats with 95 to 98% identity (like human Alu sequences) and high quality sequence data, this typically yields correct assemblies. Fast assemblies. Assemblies of cosmid- to BAC sized projects with several hundred to two thousand reads typically take only minutes to complete on high-powered workstations or personal computers. Cross_match: Fast DNA Sequence Comparisons and Vector Screening Identification of overlaps between contig ends after assembly with Phrap Identification of potential repeat sequences in assemblies. Generation of error summaries and lists after completion of sequencing projects. Estimation of vector contamination in newly created libraries.
- 14. Consed/Autofinish is a tool for viewing, editing, and finishing sequence assemblies created with phrap. Finishing capabilities include allowing the user to pick primers and templates, suggesting additional sequencing reactions to perform, and facilitating checking the accuracy of the assembly using digest and forward/reverse pair information. See the consed page for additional information. References: Gordon, David. "Viewing and Editing Assembled Sequences Using Consed", in Current Protocols in Bioinformatics,A. D. Baxevanis and D. B. Davison, eds, New York: John Wiley & Co., 2004, 11.2.1- 11.2.43.
- 15. Aligned reads window Gordon D et al. Genome Res. 1998;8:195-202 Cold Spring Harbor Laboratory Press
- 16. Navigation window. Gordon D et al. Genome Res. 1998;8:195-202 Cold Spring Harbor Laboratory Press
- 17. Trace window. Gordon D et al. Genome Res. 1998;8:195-202 Cold Spring Harbor Laboratory Press
- 18. AG-ICB-USP
- 19. AG-ICB-USP
- 20. Compare contigs window, indicating an alignment selected to investigate a contig match indicated in phrapview. Gordon D et al. Genome Res. 1998;8:195-202 Cold Spring Harbor Laboratory Press
- 21. Finishing Autofinish and manual finishing Assembly viewing/editing Consed Assembly Phrap assembled contigs - seq.fasta.screen.contigs assembly file - seq.fasta.screen.ace# Vector screening and masking Cross_Match (local alignment program) x vector.seq screened/masked file - seq.fasta.screen quality values - seq.fasta.screen.qual Conversion - phd to fasta phd2fasta.pl nucleotide sequences - seq.fasta quality values - seq.fasta.qual Quality (confidence) values assignment Phred phd files - *.phd Input chromatogram files Phred/Phrap/Consed Pipeline
- 22. Finishing Problems DNA sequencing problems a. High GC content – genomes presenting a high GC content are more prone to generate artifacts as compressions, sudden drops, bad quality regions. Try to use Dye Primer instead of Dye Terminator, change chemistry, add DMSO, increase annealing temperature, use deaza-dGTP instead of dGTP, etc. b. Palindromic regions – lead to strong secondary structures causing sudden drops. Try to use deaza-dGTP instead of dGTP, amplify the problematic region by PCR and sequence the product. c. Homopolymeric regions – can reduce DNA synthesis efficiency for some chemistries. Try to use Dye Primer instead of Dye Terminator, change chemistry (dRhodamine instead of BigDye).
- 23. DNA assembly problems a. High repeat content – highly repeated elements reduce accuracy of DNA assembly. Identify the repeat unit, screen it with Cross_Match or Repeat_Masker and mask it. Try to assemble again and add the repetitive region only at the end. Map the repetitive region using restriction enzymes to estimate its size and number of repeat units. b. High AT content – some highly biased genomes (i.e. Plasmodium falciparum; plastid genomes) can pose a problem for assembly programs. Very difficult to solve. Try to determine a restriction map and associate mapping with DNA sequencing data.
- 24. Staden Package The Staden Package was developed by Rodger Staden's group at the MRC Cambridge The main components are: pregap4 - base calling with Phred, end clipping, and vector trimming. trev - trace viewing and editing gap4 - sequence assembly, contig editing, and finishing gap5 - assembly visualisation, editing and finishing of NGS data Spin - DNA and protein sequence analysis
- 25. Staden Package PreGap Pregap is used to process raw traces It is used to mask all of the sequence such as bits of vector and poor quality sequence. Gap Gap is the Genome Assembly Program – the bit which actually assembles individual fragments into long contigs. It allows you to edit the assembly, referring back to the starting traces where they
- 27. Cufflinks assembles transcripts, estimates their abundances, and tests for differential expression and regulation in RNA- Seq samples.It can identify novel transcripts in your sequencing data by examining their alignments to the genome. Cufflinks is usually run after mapping reads to the genome with its sister tool Tophat CUFFLINKS
- 28. Submission of sequences BankIt, a WWW-based submission tool with wizards to guide the submission process Sequin, NCBI's stand-alone submission tool with wizards to guide the submission process is available by FTP for use on for MAC, PC, and UNIX platforms. tbl2asn, a command-line program, automates the creation of sequence records for submission to GenBank using many of the same functions as Sequin. It is used primarily for submission of complete genomes and large batches of sequences and is available by FTP for use on MAC, PC and Unix platforms. Submission Portal, a unified system for multiple submission types. Currently only 16S ribosomal RNA from uncultured bacteria/archaea can be submitted with the GenBank component of this tool. This will be expanded in the future to include other types of GenBank submissions. Genome and Transcriptome Assemblies can be submitted through the WGS and TSA portals, respectively. Barcode Submission Tool, a WWW-based tool for the submission of sequences and trace read data for Barcode of Lifeprojects based on the COI gene.
- 29. BankIt, Submission Portal and Barcode Submission Tool entries are automatically submitted to GenBank. Submissions made with Sequin or tbl2asn must be mailed to gb-sub@ncbi.nlm.nih.gov. Large files which may be truncated during mailing with conventional mail tools should be submitted directly using Sequin MacroSend. Submissions of Raw Sequence Reads Reads of Sanger-style sequencing can be submitted to the Trace Archive. Runs of next-generation sequencing, for example from 454 or Illumina, can be submitted to the Sequence Read Archive (SRA).
- 30. You should use BankIt if: You have a single sequence, a simple set of sequences (for example:16S rRNA, matK, ITS/rRNA, amoE, tefB, cytb, or COI sets), or a small batch of different sequences You prefer to use a web-based submission tool The feature annotation for your sequences is not complicated You do not require advanced sequence analysis tools
- 31. Sequin
- 32. You should use tbl2asn if: Your sequence has a lot of annotation You are submitting a large batch of sequences You have Whole Genome Shotgun (WGS) submissions You have complete genome submissions You are submitting FLIC sequences
- 33. Sequence formats sequence formats.docx NCBI Gen Bank EMBL Stanford University
Editor's Notes
- Aligned reads window, in color means quality and tags color mode. The top line gives the contig sequence, and below it are the read sequences for the top strand (right-pointing arrows) and bottom strand (left-pointing arrows). Read names are in yellow. The background gray scale indicates base quality, with the highest quality being white and the lowest quality black. Red indicates a character (such as the x or *) that disagrees with the contig sequence. (x) A base that has been masked by cross-match as being vector. (*) A pad that is inserted by phrap to align reads that have insertions and deletions. Tags are indicated by colored bars covering the bottom half of the background square for each base. The blue tag represents sequencing vector, and the orange tag indicates compressions. An edited base has a green tag attached. Gray letters on a black background indicate that phrap clipped these bases off because of low quality.
- Navigation window. Each line contains the contig name, the read name, the range of consensus positions, and an indication of the problem. The Go, Prev, and Next buttons cause the associated aligned reads window to scroll to the location on the currently highlighted line, the line above it, or the line below it, respectively. All items can be visited in order by repeatedly clicking Next. The Save button creates a file containing the list.
- Trace window. The lines in each panel above the trace chromatogram indicate the following: (con) consensus position; (rd) read position; (con) consensus bases; (edt) editable read bases; (phd) phredbase calls; and (ABI) ABI base calls. The H and V scale bars change the horizontal and vertical magnification of the traces. (Scroll together Yes/No buttons) Allows the user to scroll the traces individually or locked together. (Remove) Removes this trace panel from the window. (Undo) Undo the last edit operation on that read.
- Compare contigs window, indicating an alignment selected to investigate a contig match indicated in phrapview. (Top): Aphrapview window showing matches between contigs as red lines. (Bottom) consed compare contigs window showing the sequence alignment of one match from the phrapview window. The two rows of bases in lowercase are the unaligned contigs, which can be scrolled relative to each other. The two rows of bases in uppercase are the aligned contigs, which are locked together if they are scrolled. (x) A mismatch. Red cursors indicate the bases to be pinned together. (Align button) Click this to show alignment. (Scroll Top Contig/Scroll Bottom Contig buttons) After clicking on a base, this causes the Aligned reads window to scroll to the appropriate location.