Download as PDF, PPTX








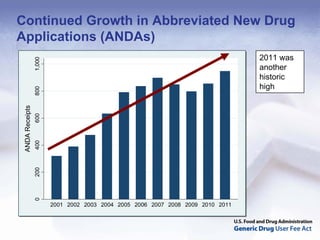
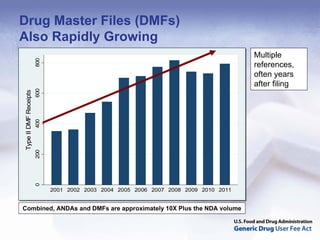
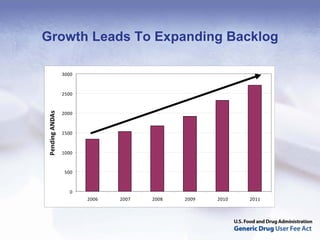
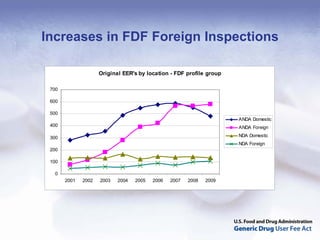
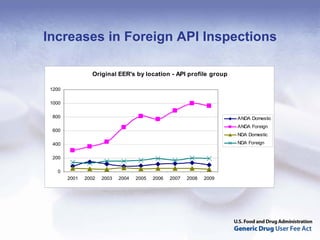























The document outlines the process and goals of a new act related to generics. It discusses the large and growing generic drug industry and FDA's challenges in regulating it given expanding volume. The act would authorize $299 million annually in user fees to fund a 10-month review cycle for ANDAs and goals for inspections. Metrics include hiring staff, reducing the ANDA backlog, timelines for reviews and amendments, and controlled correspondence. The goals aim to gain efficiencies through electronic submissions, risk-based inspections, and leveraging third-party regulators.





























































































