Downloaded 70 times



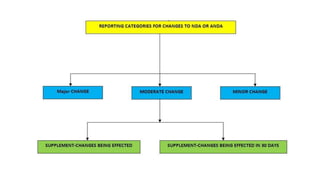













































The document discusses post-approval changes that can be made to approved NDAs and ANDAs. It describes four reporting categories for post-approval changes based on their potential impact: major changes requiring prior approval, moderate changes reported via a CBE-30 or CBE-0 supplement, minor changes reported annually. Examples are provided for different types of changes that fall under each reporting category. The levels of reporting ensure that manufacturers can make certain changes while providing appropriate notification to the FDA depending on the level of change.













































































