Downloaded 306 times
![ABBREVIATED NEW DRUG APPLICATION
[ANDA]
Mr. Sagar Kishor savale
[Department of Pharmaceutics)]
avengersagar16@gmail.com
2015-2016
1](https://image.slidesharecdn.com/anda-160709072040/85/ANDA-1-320.jpg)












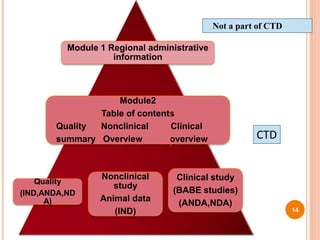
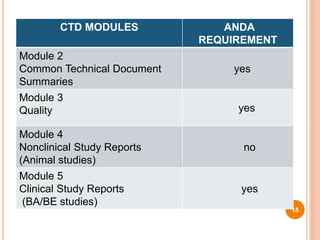


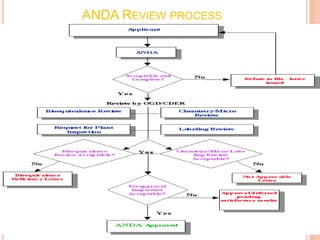
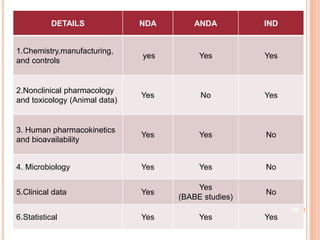







The document provides information about abbreviated new drug applications (ANDAs), which are designed to allow approval of generic drug products that are equivalent to already approved brand name drugs. An ANDA must show a generic drug is comparable to the reference drug in dosage form, strength, quality and performance. It does not require preclinical and clinical trials but must demonstrate bioequivalence through bioavailability and bioequivalence studies. The ANDA contents and review process are outlined according to the Common Technical Document format in five quality, nonclinical, and clinical modules.








![Abbreviated New Drug Application [ANDA]](https://cdn.slidesharecdn.com/ss_thumbnails/abbreviatednewdrugapplicationanda-160619062810-thumbnail.jpg?width=640&height=640&fit=bounds)




















![Investigational New drug application [INDA]](https://cdn.slidesharecdn.com/ss_thumbnails/investigationalnewdrugapplicationinda-160619063044-thumbnail.jpg?width=640&height=640&fit=bounds)













































































