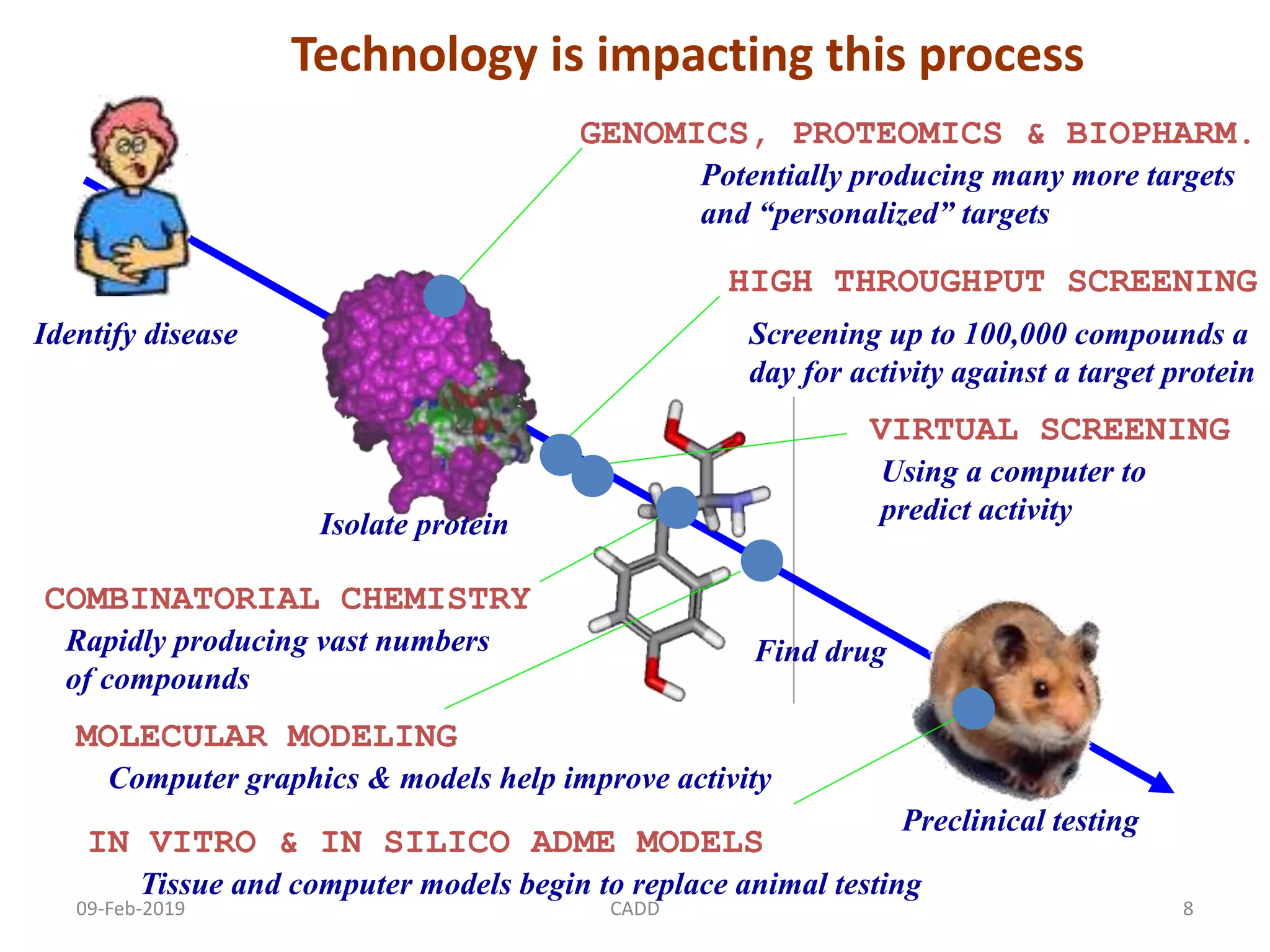
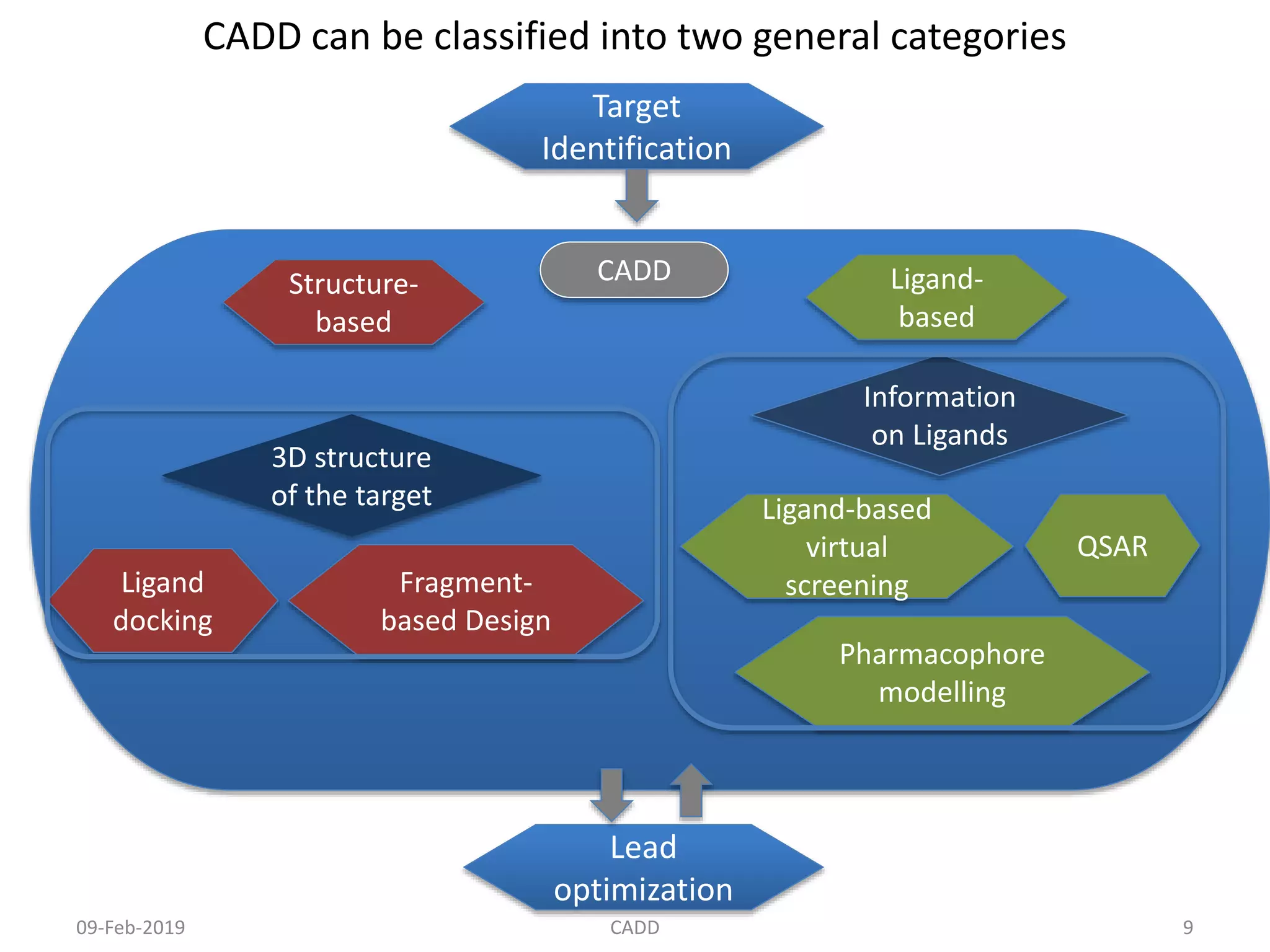
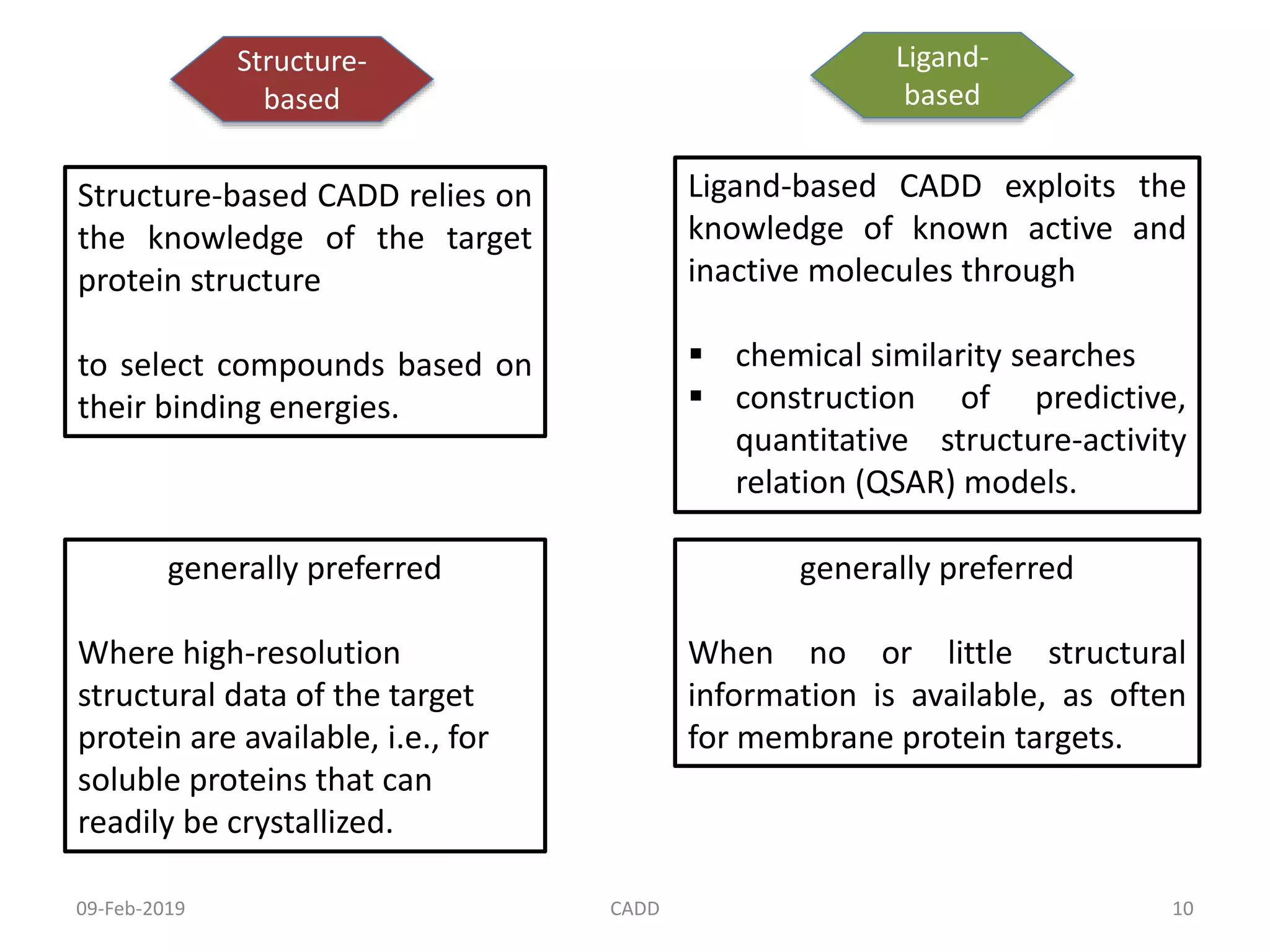
The document discusses computer-aided drug design (CADD) and its role in drug discovery. CADD uses computer software and modeling to aid the drug design process and identify new drug candidates. It reduces the time and cost of drug discovery compared to traditional methods. Some of the earliest approved drugs discovered using CADD include Dorzolamide, Captopril, and drugs for HIV. CADD approaches include structure-based design using protein structure data and ligand-based design using information about known active/inactive ligands. Key steps involve target identification, obtaining protein structures, ligand docking simulations, and lead optimization.














![Lipophilic parameters
Lipophilicity is partitioning of the compound between
an aqueous and non-aqueous phase.
Partition coefficient:
• P = [drug] in octanol / [drug] in water
Typically over a small range of log P, e.g. 1-4, a straight
line is obtained
e.g. log 1/C = 0.75 log P + 2.30
• If graph is extended to very high log P values, then
get a parabolic curve
log 1/C = - k1 (log P)2 + k2 log P + k3
• When P small, dominated by log P term
• When P large, log P squared dominates & so activity
decreases
09-Feb-2019 CADD 18](https://image.slidesharecdn.com/knp-cadd-190730093054/75/COMPUTER-AIDED-DRUG-DESIGN-18-2048.jpg)




























































































