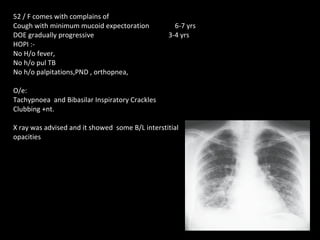
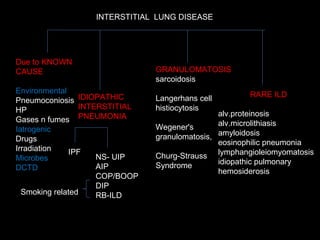
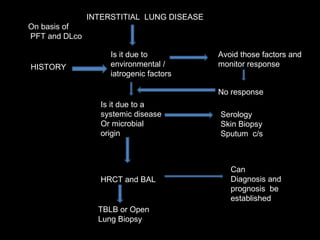
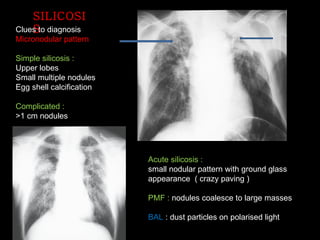
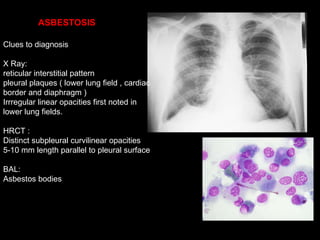
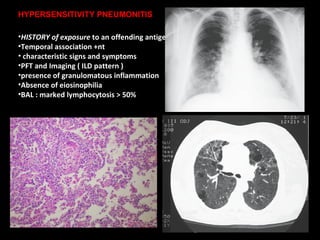
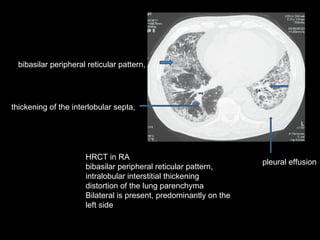
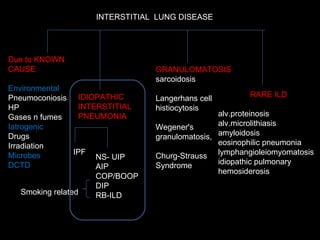
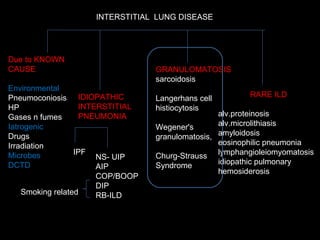
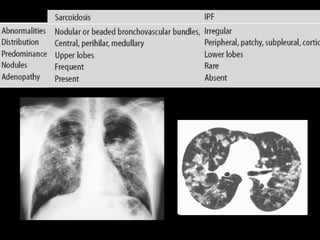
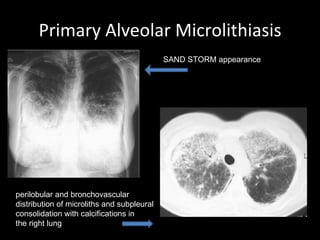
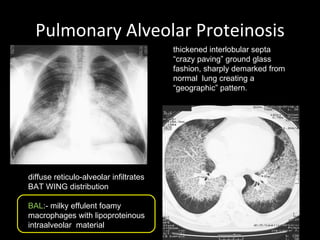
The document discusses interstitial lung disease (ILD), including its common features, types, causes, diagnostic approach and treatment. It describes various ILD types such as idiopathic pulmonary fibrosis and sarcoidosis. Imaging and biopsy are used to diagnose ILD and determine prognosis. Treatment involves identifying and removing environmental causes, suppressing inflammation, and managing complications like right heart failure.








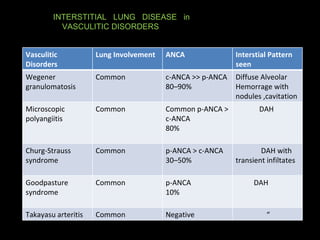
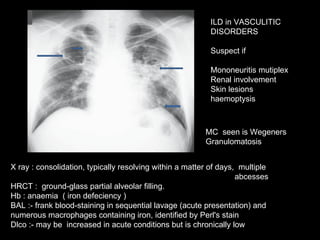

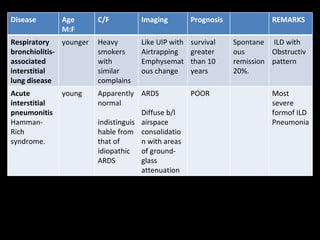
![Disease Age M:F C/F Imaging Prognosis REMARKS Nonspecific interstitial pneumonitis (NSIP) 40-50 May be indistinguishable from UIP Like But uniform in time, suggesting response to single injury UIP Honeycombing is rare. Prognosis good but depends on the extent of fibrosis at diagnosis greater than 10 years. But Surgical Biopsy is needed to confirm. Cryptogenic organizing pneumonitis (bronchiolitis obliterans organizing pneumonia [BOOP]) 50–60 Abrupt onset, frequently weeks to a few months following a flu-like illness. constitutional symptoms are common Ground glass infiltrate subpleural consolidation and bronchial wall thickening and dilation. Xray – interstitial pattern with nodules Good Rule out infection and treat with steroids](https://image.slidesharecdn.com/interstitiallungdisease-100122140036-phpapp01/85/Interstitial-Lung-Disease-22-320.jpg)
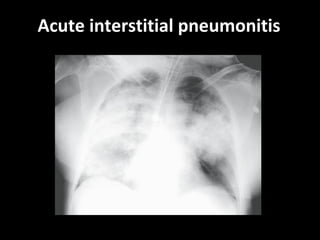
![Cryptogenic organizing pneumonitis (bronchiolitis obliterans organizing pneumonia [BOOP])](https://image.slidesharecdn.com/interstitiallungdisease-100122140036-phpapp01/85/Interstitial-Lung-Disease-25-320.jpg)














![Interstitial Lung Diseases [ILD] Approach to Management](https://cdn.slidesharecdn.com/ss_thumbnails/interstitiallungdiseases-arunvasireddy-19october2015-seminar-171016041856-thumbnail.jpg?width=640&height=640&fit=bounds)





































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)








