Downloaded 102 times




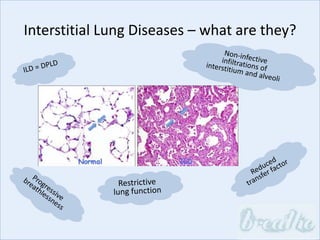
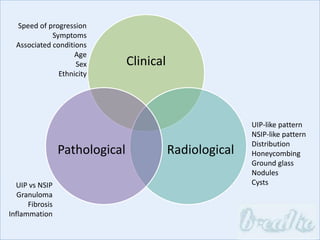
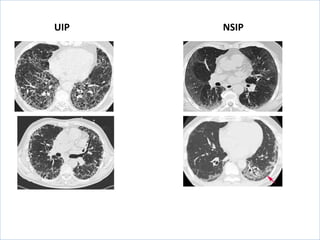
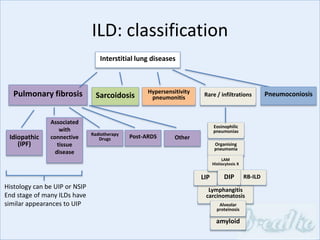


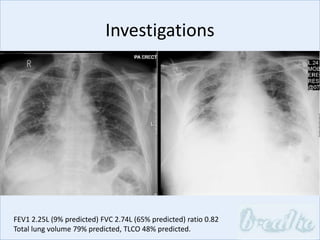
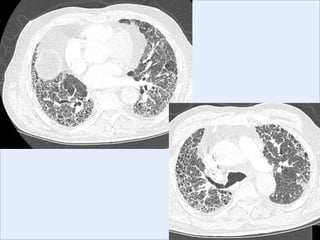


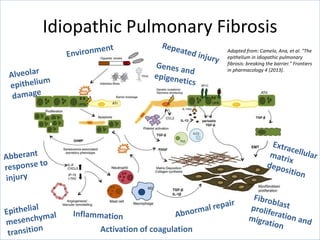
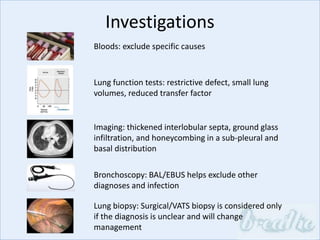
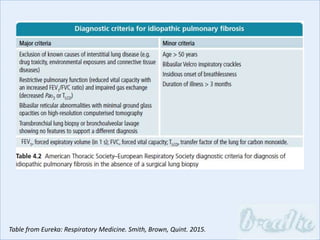




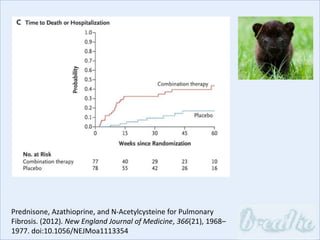






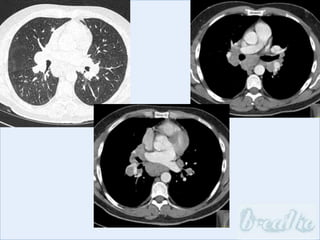
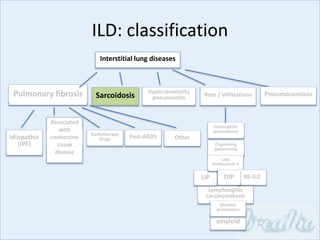


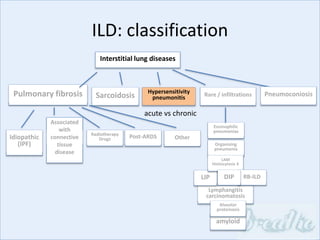

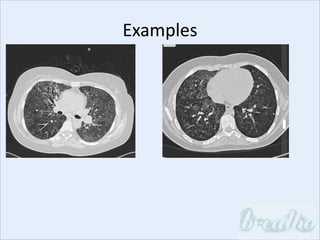
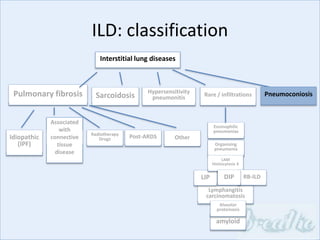

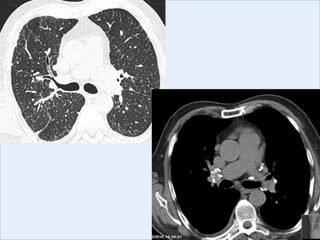

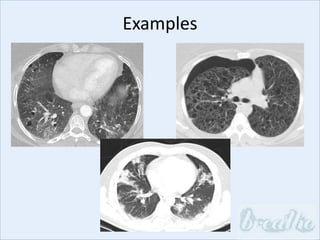








The document discusses interstitial lung diseases (ILDs), including: - ILDs are a group of non-infectious diseases affecting the lungs' interstitium and alveoli. - Classification is based on clinical, radiological, and pathological features. Idiopathic pulmonary fibrosis (IPF) and non-specific interstitial pneumonia (NSIP) are examples. - Diagnosis involves clinical history, imaging like CT, lung function tests, and sometimes biopsy. Prognosis and treatment vary depending on the specific ILD.



































































































