1. Interstitial lung diseases (ILDs) involve the lung parenchyma including the alveoli, capillaries, and tissues between them.
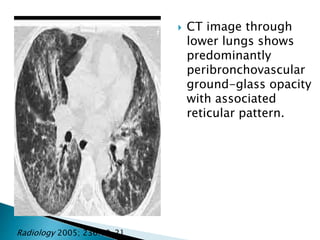
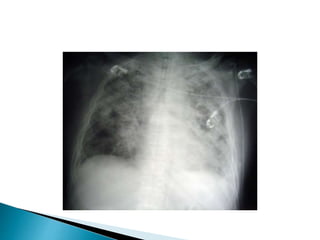
2. Patients commonly present with progressive dyspnea, cough, and interstitial opacities on imaging.
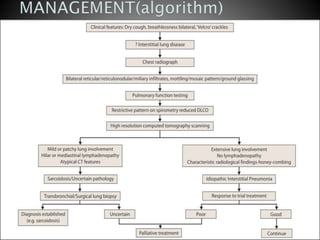
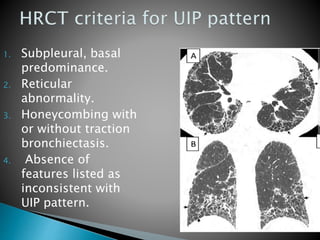
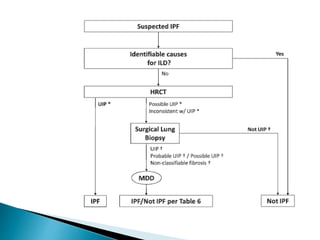
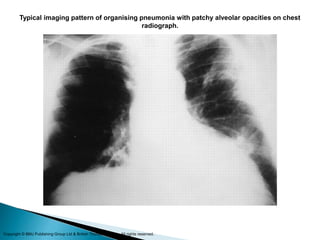
3. A thorough evaluation includes pulmonary function tests, imaging, biopsy, and ruling out other known causes to identify the underlying ILD.





















































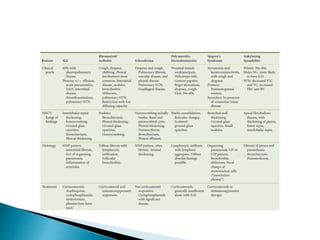










![Interstitial Lung Diseases [ILD] Approach to Management](https://cdn.slidesharecdn.com/ss_thumbnails/interstitiallungdiseases-arunvasireddy-19october2015-seminar-171016041856-thumbnail.jpg?width=640&height=640&fit=bounds)


































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)















