Download as PDF, PPTX





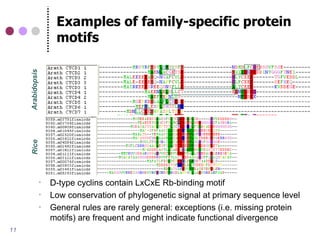
![Examples of family-specific protein
motifs
B-type cyclins have HxKF signature
Cyclin destruction boxes (B1-type cyclin R-[AV]LGDIGN)
10](https://image.slidesharecdn.com/bitscompgenomics201102genefamilyanalysis-120116040620-phpapp02/85/BITS-Comparative-genomics-gene-family-analysis-10-320.jpg)




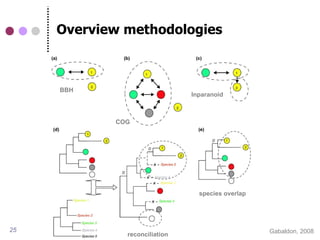
![Advanced methods for protein
(orthology) clustering
Sequence similarity-based
COG (RBH) [Tatusov 1997]
InParanoid [Remm et al., 2001]
Tribe-MCL [Van Dongen 2000]
OrthoMCL [Li et al., 2003]
Phylogenetic tree-based
PhylomeDB [Huerta-Cepas et al., 2007]
Ensembl Compara [Vilella et al., 2008]
24](https://image.slidesharecdn.com/bitscompgenomics201102genefamilyanalysis-120116040620-phpapp02/85/BITS-Comparative-genomics-gene-family-analysis-24-320.jpg)




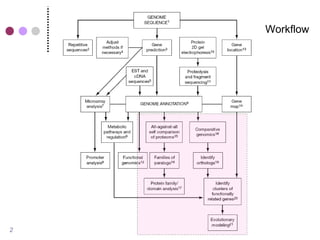
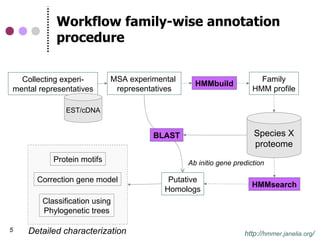
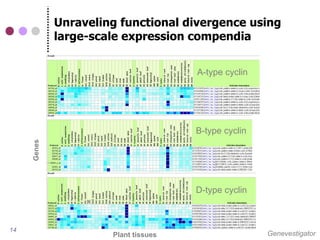
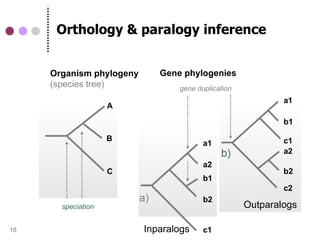
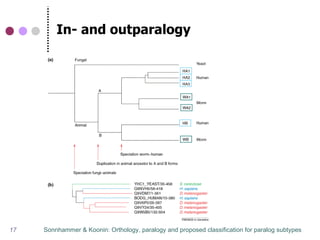
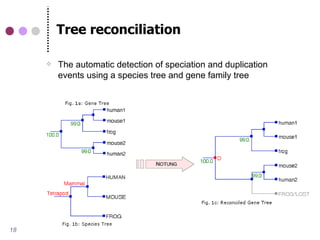
This document discusses comparative genomics and gene family analysis. It describes clustering proteins into families to study evolution, orthology, paralogy, gene duplication and loss. Gene family analysis allows detection of errors in gene structure annotation by comparing sequences. Phylogenetic trees classify gene families and reveal functional divergence. Resources for orthology analysis include Ensembl, OrthoMCLDB and YGOB. The goal of hands-on analysis is to characterize the talin 2 gene family.