Downloaded 11 times


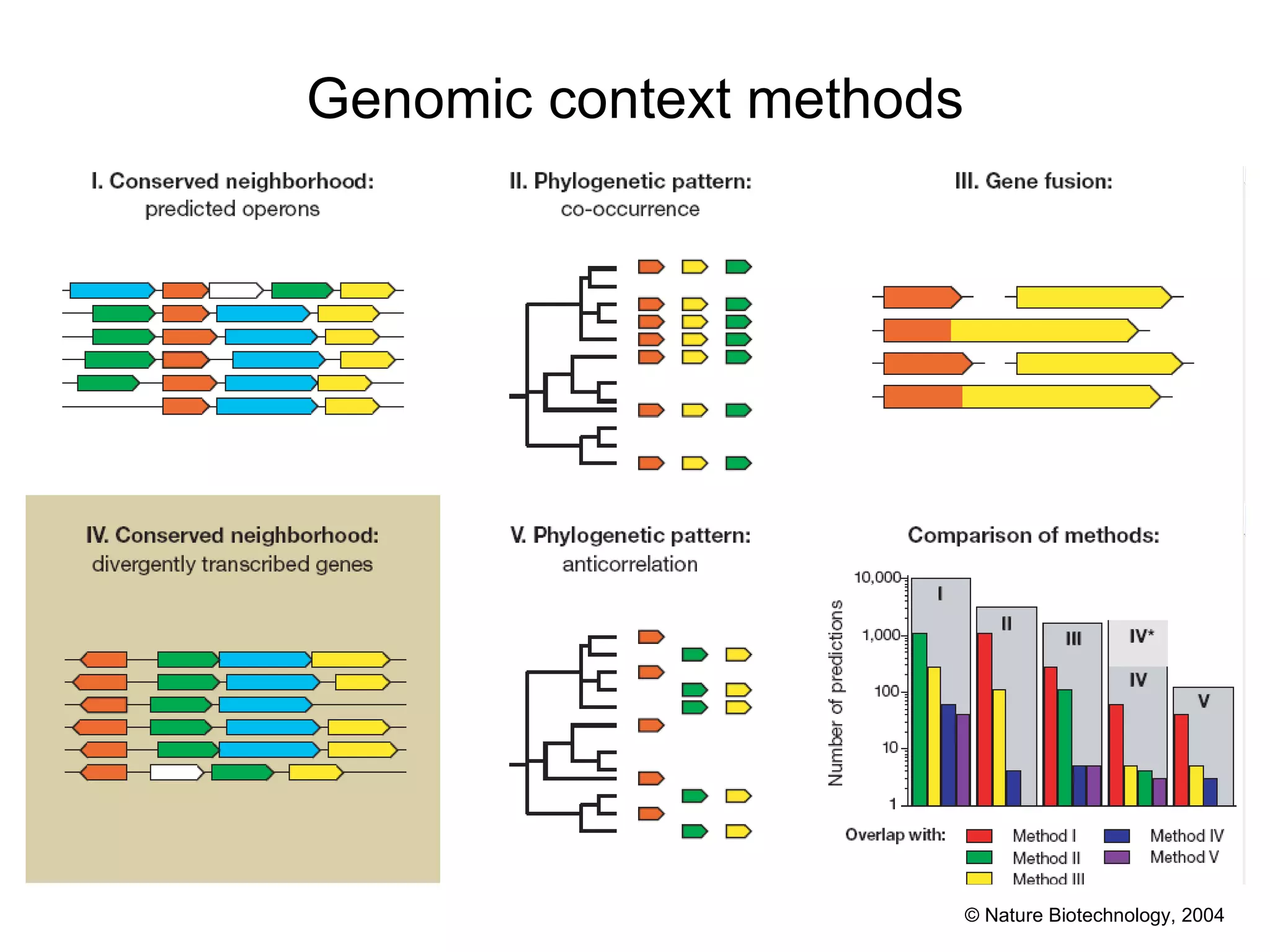



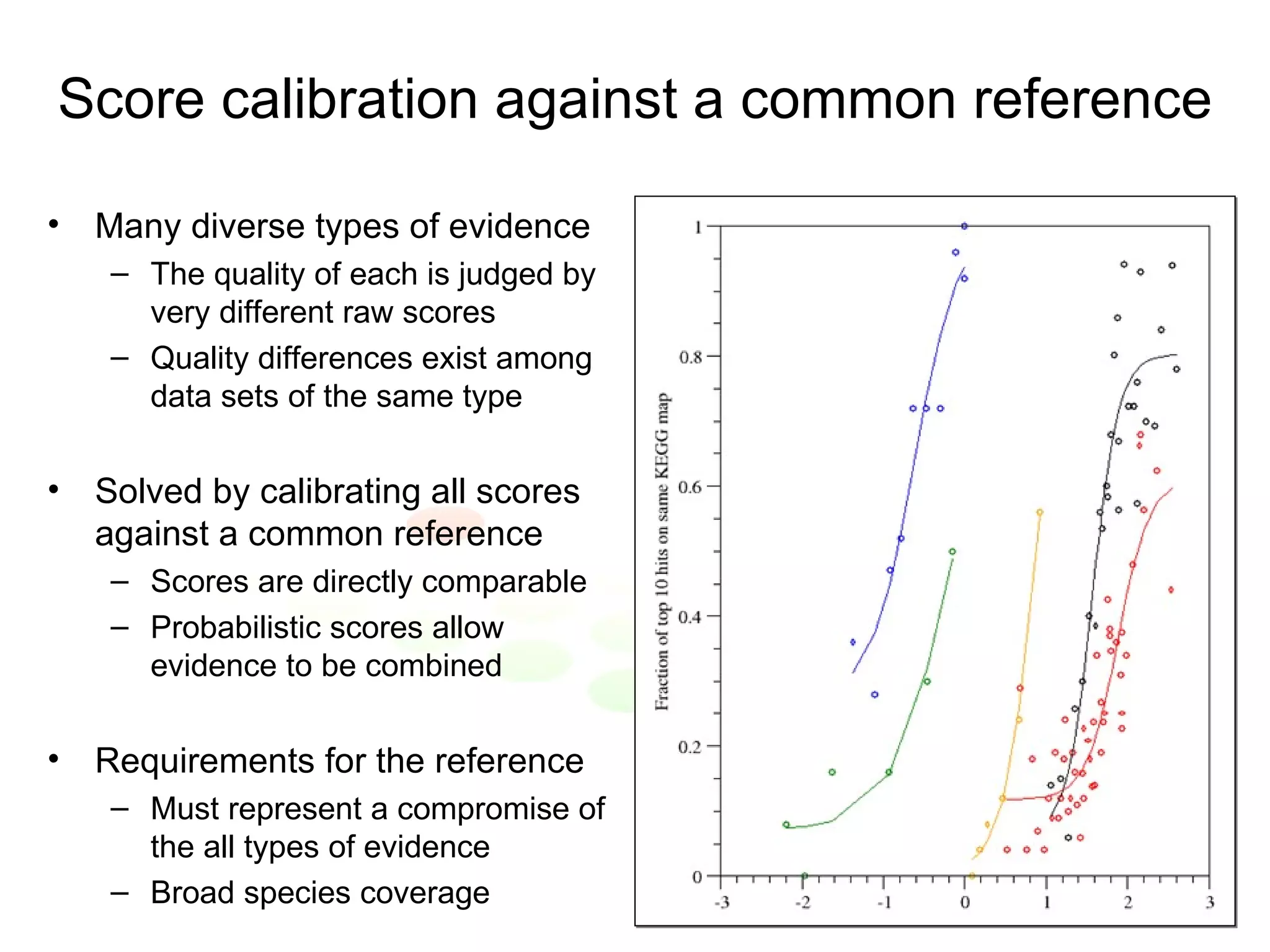







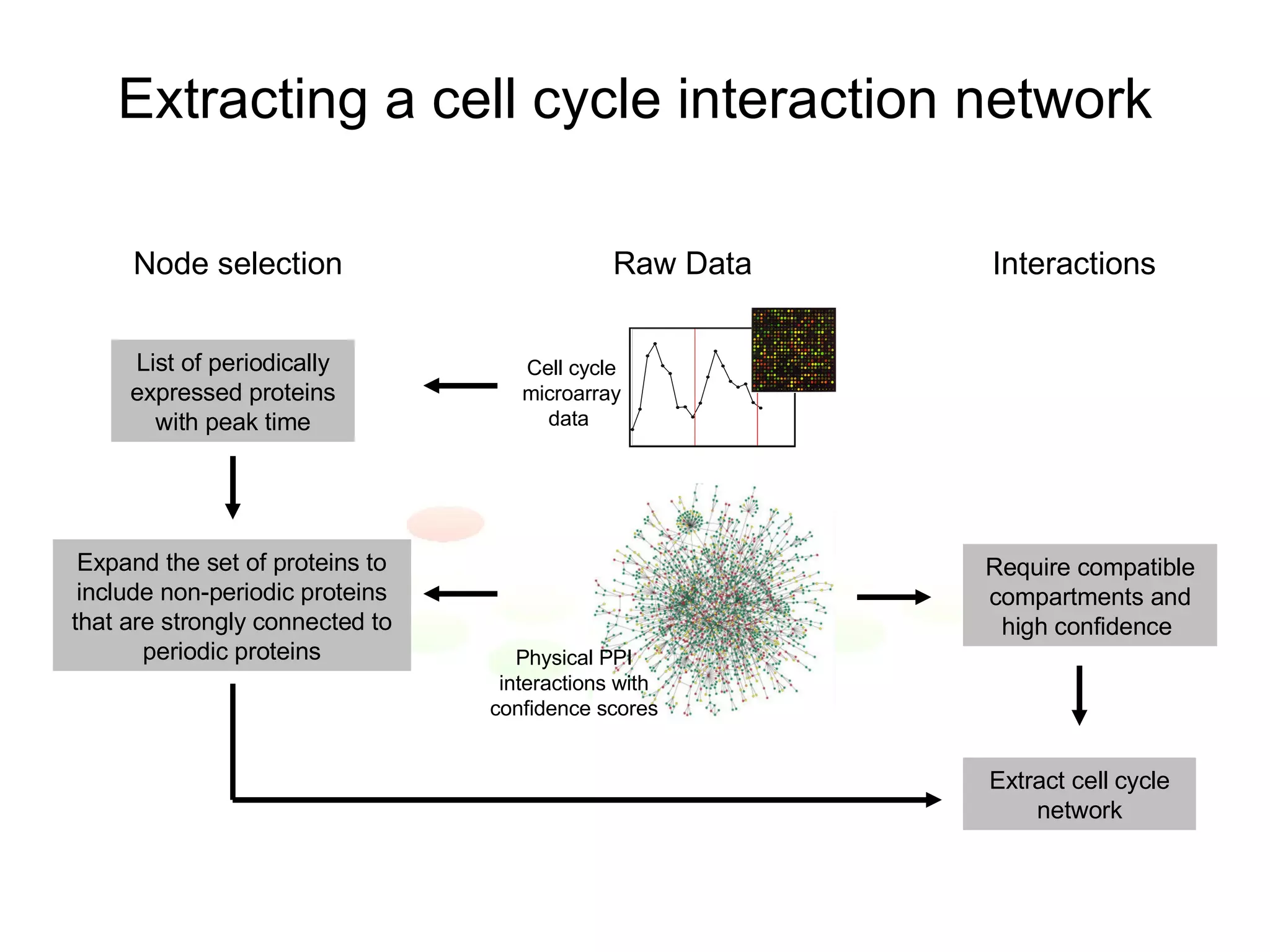

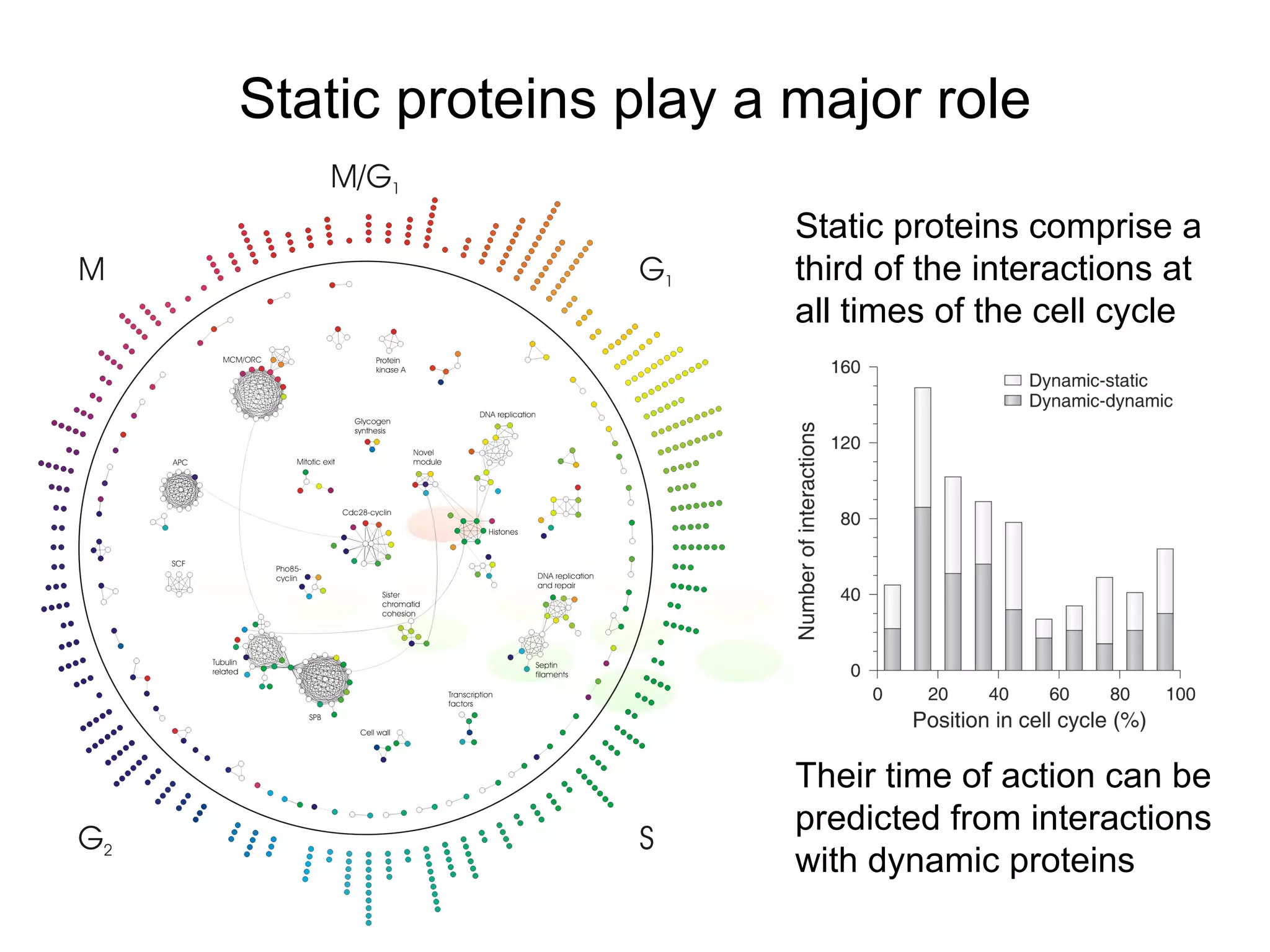
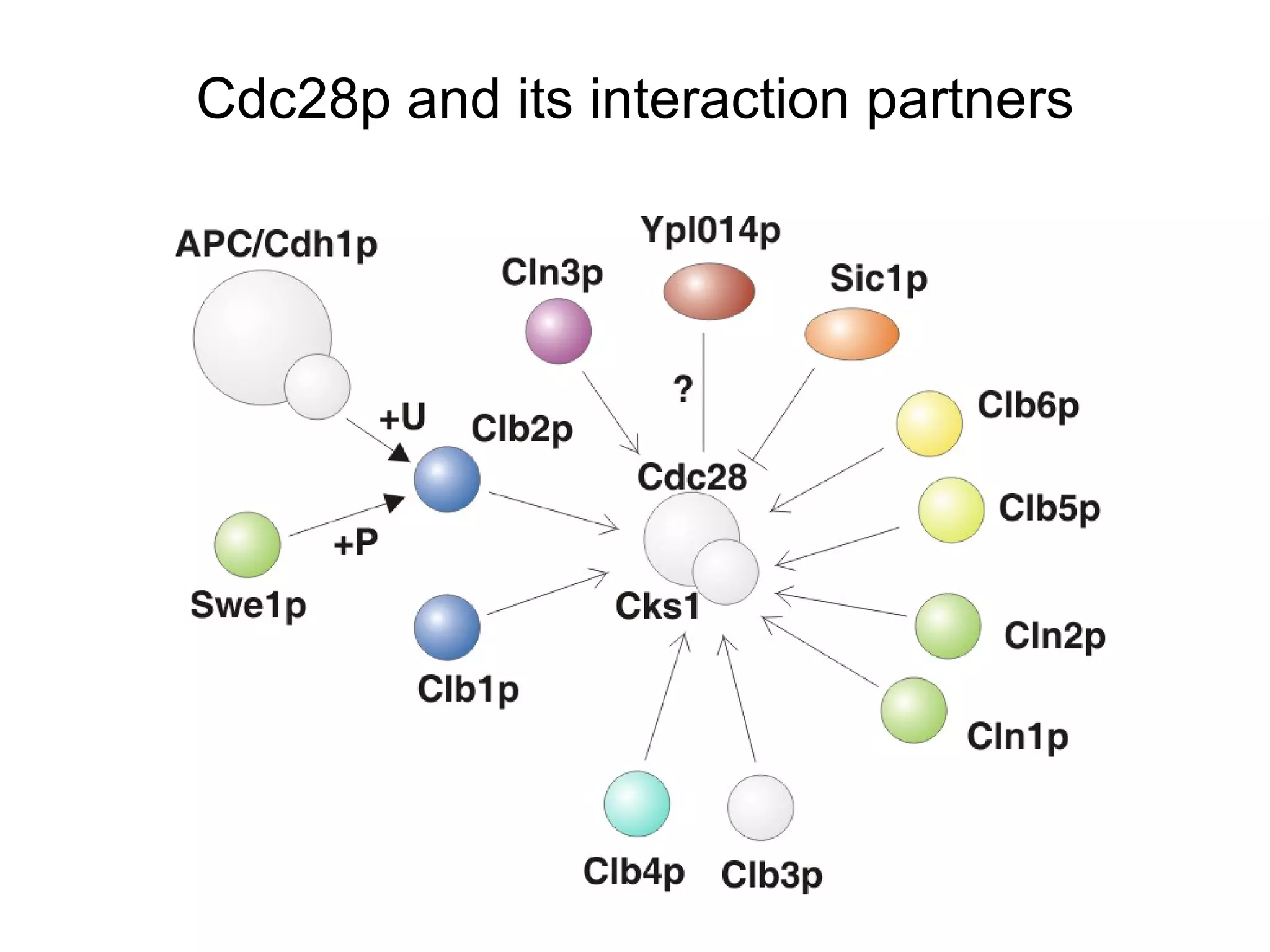
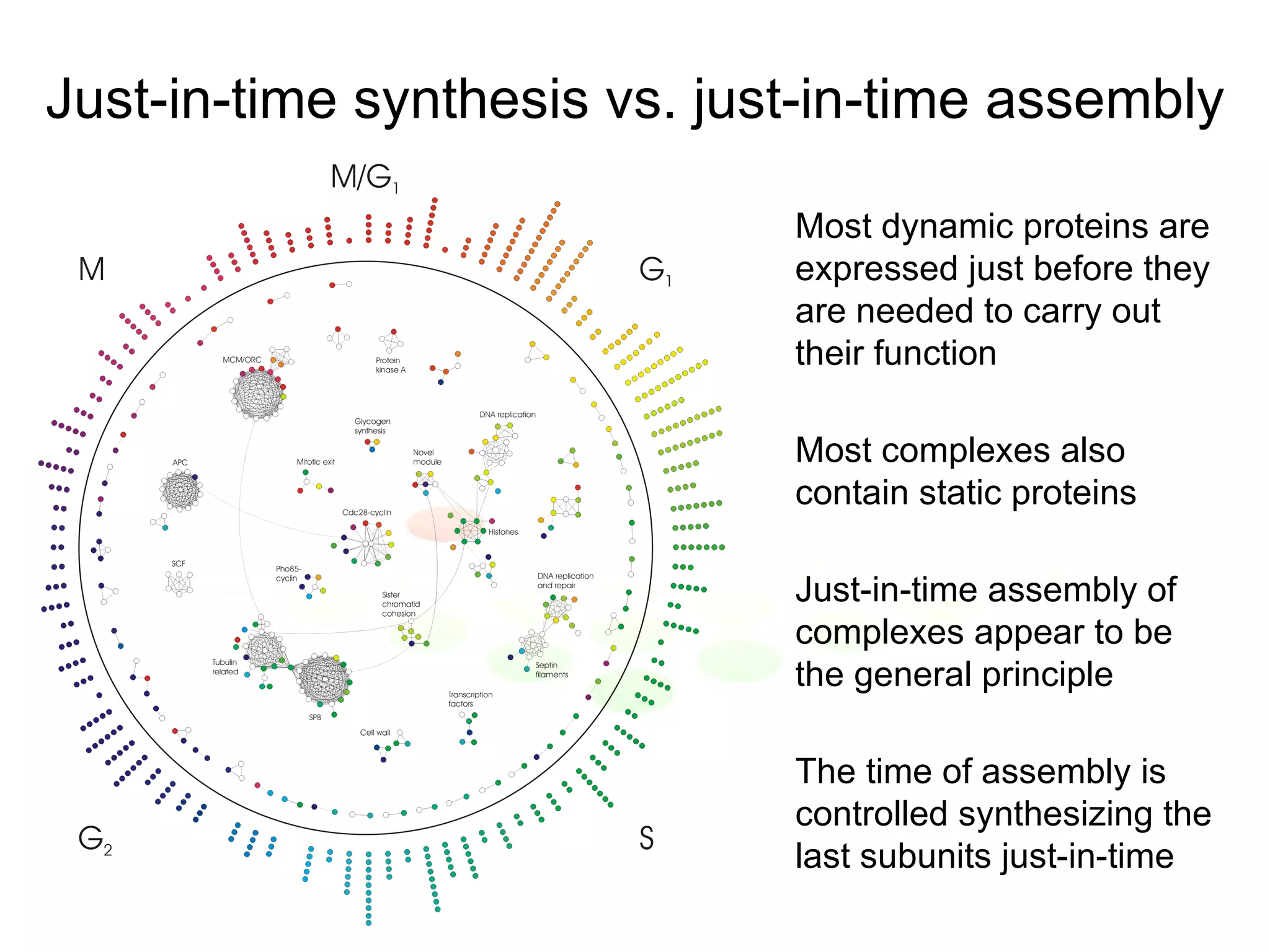
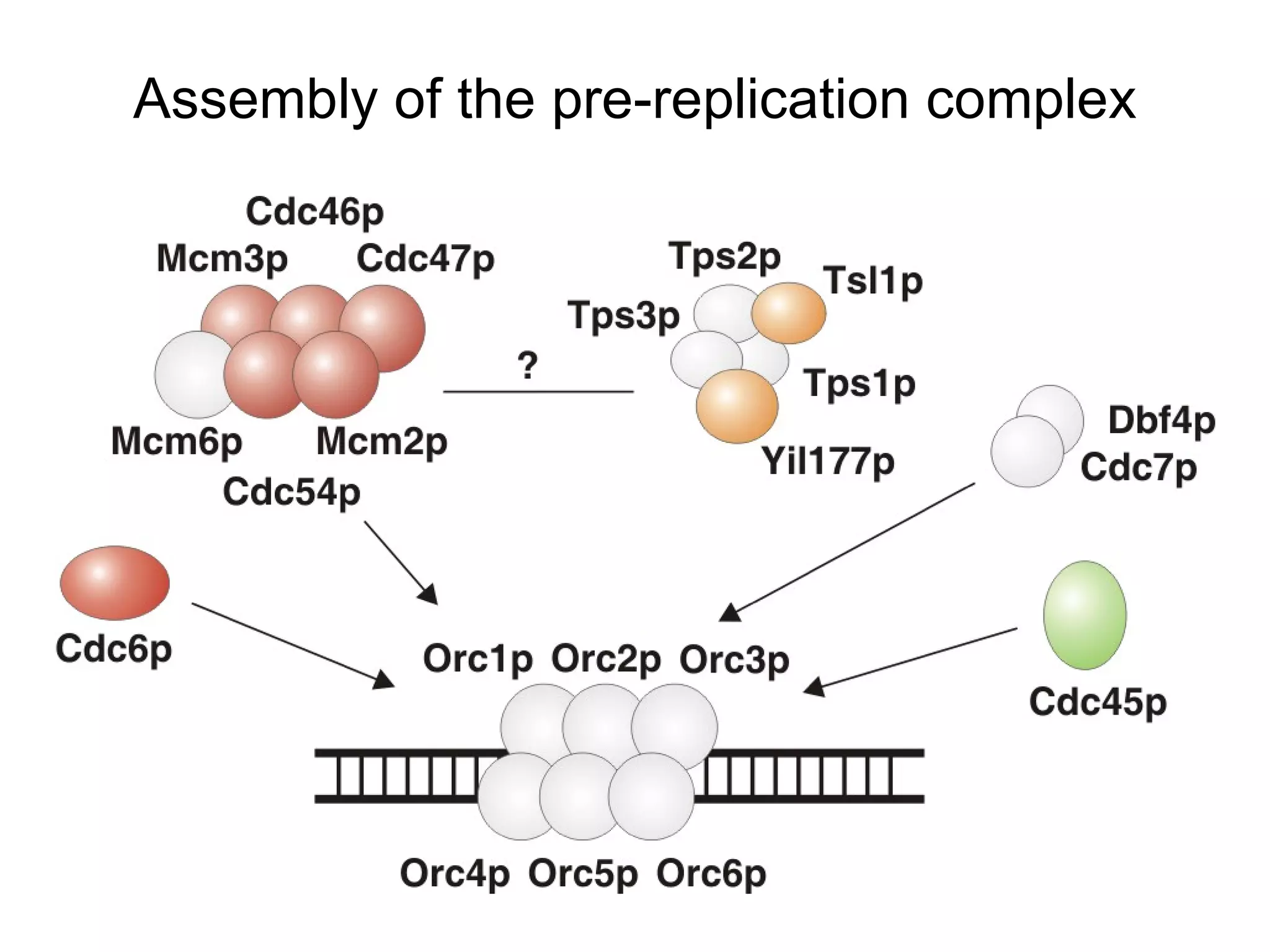
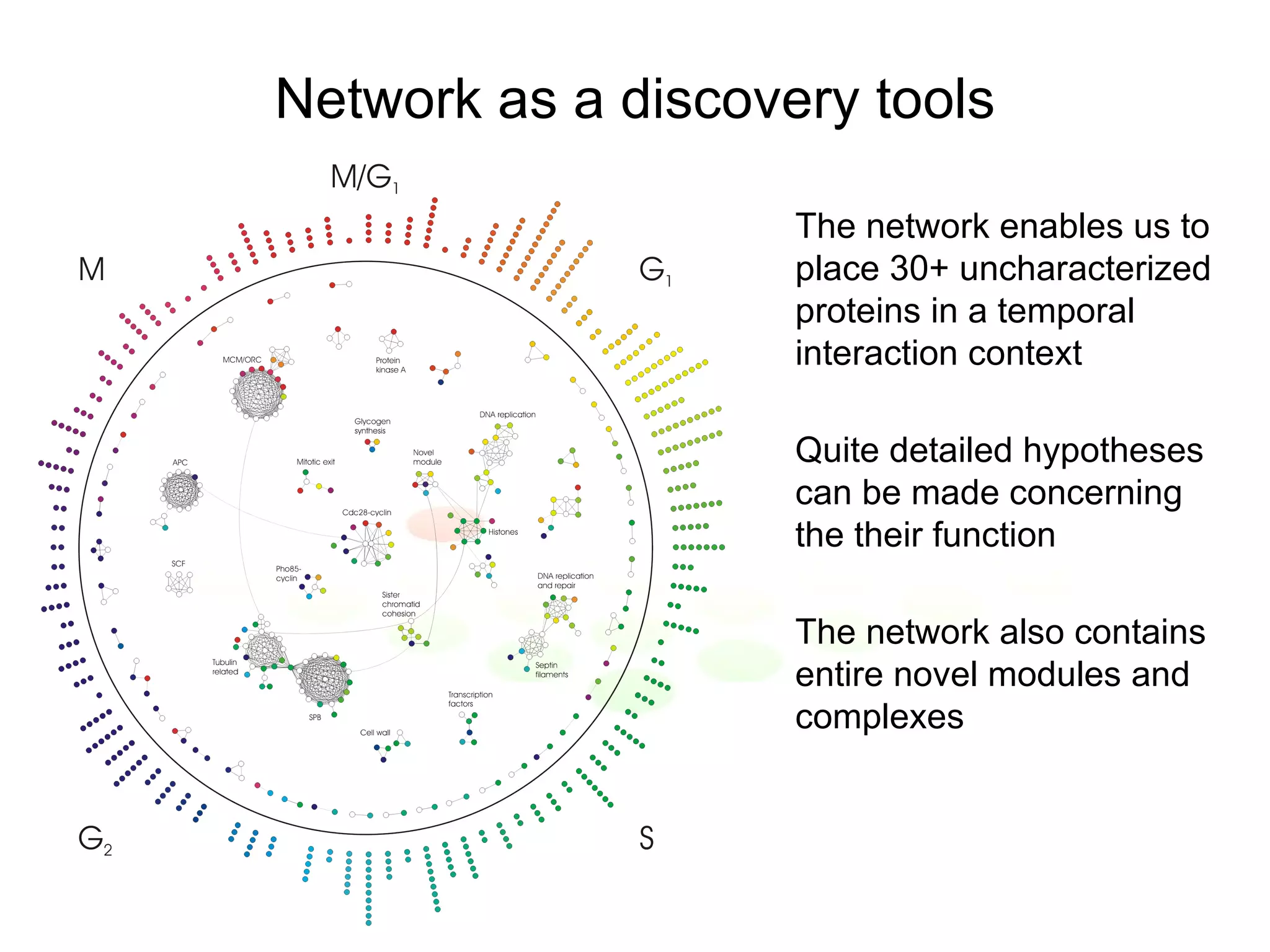
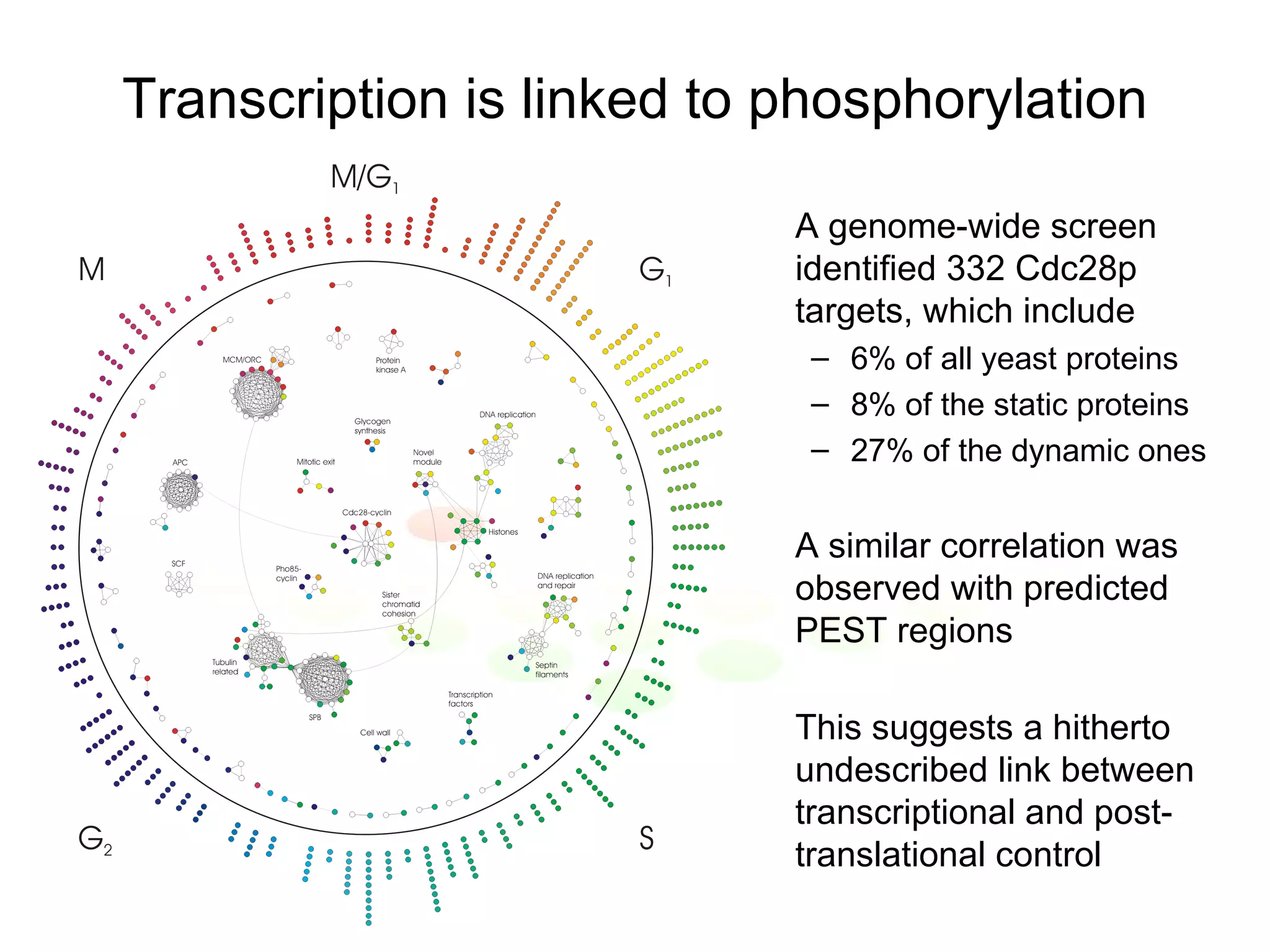




The document discusses methods for integrating diverse evidence like genomic context, gene fusions, expression data, and protein-protein interaction screens to infer functional associations between proteins across species. It describes benchmarking different data sources against a common reference to make scores comparable and combining evidence probabilistically. The methods are applied to construct an accurate protein interaction network for the yeast cell cycle by extracting a periodically expressed subset and integrating temporal expression and interaction data.