This document provides an outline and introduction to dissolution studies. It discusses what dissolution is, why dissolution studies are important, and factors that can affect dissolution. It also covers different dissolution models, including zero order, first order, Higuchi, and diffusion layer models. The models are used to understand drug release kinetics and develop in vitro-in vivo correlations. Dissolution studies are important tests used to evaluate drug release from dosage forms and ensure batch-to-batch quality and bioavailability.





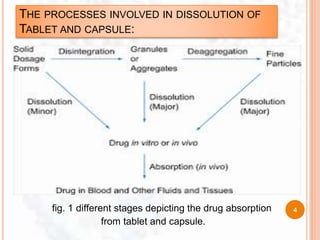




![ Dissolution profile:
It is a graphical representation [in term of
concentration vs time ] of complete release of A.P.I. from a
dosage form in an appropriate selected dissolution medium.
i.e. in short it is a measure of release of A.P.I. from dosage
form with respect to time.
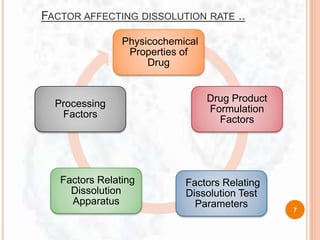
IT’s NEED:
To develop invitro- invivo correlation which can help to
reduce costs, speed up product development.
To stabilize final dissolution specification for
pharmacological dosage form.
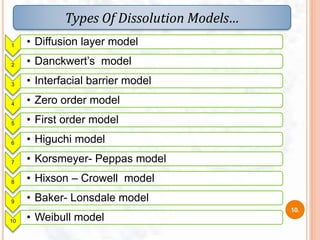
DISSOLUTION MODEL’s AND IT’s NEED
9](https://image.slidesharecdn.com/dissolutionmodelssem1-170508105530/85/Dissolution-models-sem-1-11-320.jpg)


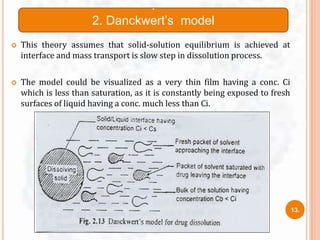


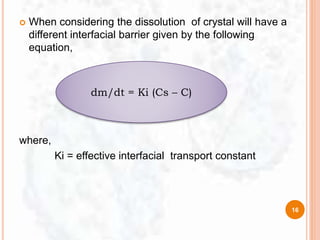

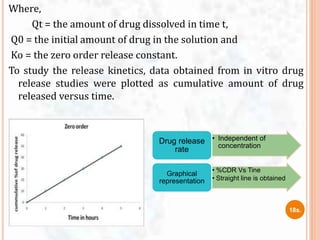

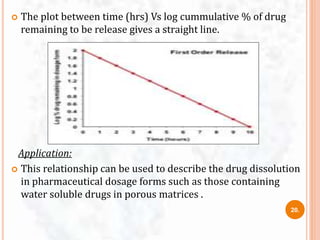

![ The study of dissolution from a planar system having a
homogeneous matrix can be obtained by the equation:
Where,
A = amount of drug released in time ‘t’ per unit area
D = diffusivity of drug molecule in the matrix substance
C = initial drug concentration
Cs = drug solubility in the matrix media.
A=[D(2C-Cs)Cs X t]1/2
22.](https://image.slidesharecdn.com/dissolutionmodelssem1-170508105530/85/Dissolution-models-sem-1-24-320.jpg)
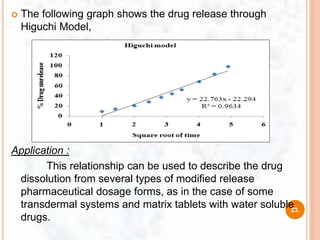

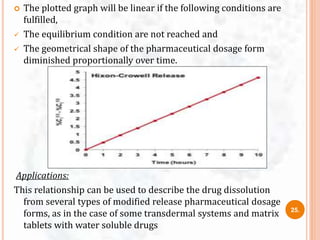

![ This model was developed by Baker and Lonsdale (1974)
from the Higuchi model and described the drug release from
spherical matrices by using the equation:
Where,
At = drug released amount at time t
A∞ = amount of drug released at an infinite time,
Dm = diffusion coefficient,
Cms = drug solubility in the matrix,
ro = radius of the spherical matrix
Co = initial concentration of drug in the matrix
9. Baker- Lonsdale model
f1= 3/2[1-(1-Ct/C∞)2/3] Ct/C∞ = (3DmCms)/(ro2Co)X t
28.](https://image.slidesharecdn.com/dissolutionmodelssem1-170508105530/85/Dissolution-models-sem-1-30-320.jpg)
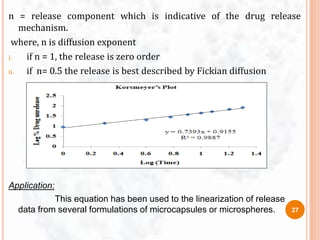
![ To study the release kinetics, data obtained from in vitro drug
release studies were plotted as [d (At / A∞)] / dt with respect
to the root of time inverse.
Application:
This equation has been used to the linearization of release
data from several formulations of microcapsules or
microspheres.
29.](https://image.slidesharecdn.com/dissolutionmodelssem1-170508105530/85/Dissolution-models-sem-1-31-320.jpg)
![ Wiebull model is generally applied to drug dissolution or release from
pharmaceutical dosage forms
These accumulated fraction of drug M in solution at time t is given by
Wiebull equation,
Where,
m = % dissolved in time ‘t’
a= scale parameter which defines the time scale of the dissolution
process
Ti = lag time( generally zero)
b = shape factor
What is the need of this equation?
It can be widely used for analysis and characterisation
Of of Drug Dissolution process from different dosage form.
10. Wiebull Model
M= Mo[1-e-(t-T/a)b]
30.](https://image.slidesharecdn.com/dissolutionmodelssem1-170508105530/85/Dissolution-models-sem-1-32-320.jpg)

![Mathematical models for drug release or drug dissolution:
Sr.No. Model Mathematical
equation
Release Mechanism
1 Zero order C=Co-Kot Diffusion Mechanism
2 First order dc/dt = K (CS - C) Fick‟s first law,
diffusion Mechanism
3 Higuchi Model A=[D(2C-Cs)Cs X
t]1/2
Diffusion medium
based Mechanism in
Fick‟s first law
4 Korsemeyer- Peppas
Model
Mt / M∞ = Ktn Peppas Model
Ct/C∞=Ktn Semi empirical
model, diffusion based
mechanism
5 Hixson–Crowell Model Mo1/3 - Mt1/3 = κ t Erosion release
mechanism
6 Weibull Model M= Mo[1-e-(t-T/a)b] Empirical model ,life-
time distribution
function
7 Baker–Lonsdale
Model
f1=3/2[1-(1-
Ct/C∞)2/3] Ct/C∞=Kt
Release of drug from
spherical matrix
32.](https://image.slidesharecdn.com/dissolutionmodelssem1-170508105530/85/Dissolution-models-sem-1-34-320.jpg)































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)








