Downloaded 68 times

![Dr.G.Bhanu Prakash – www.gims-org.com @ Global institute of Medical sciences – The Medical Biochemistry Tit-Bits
3
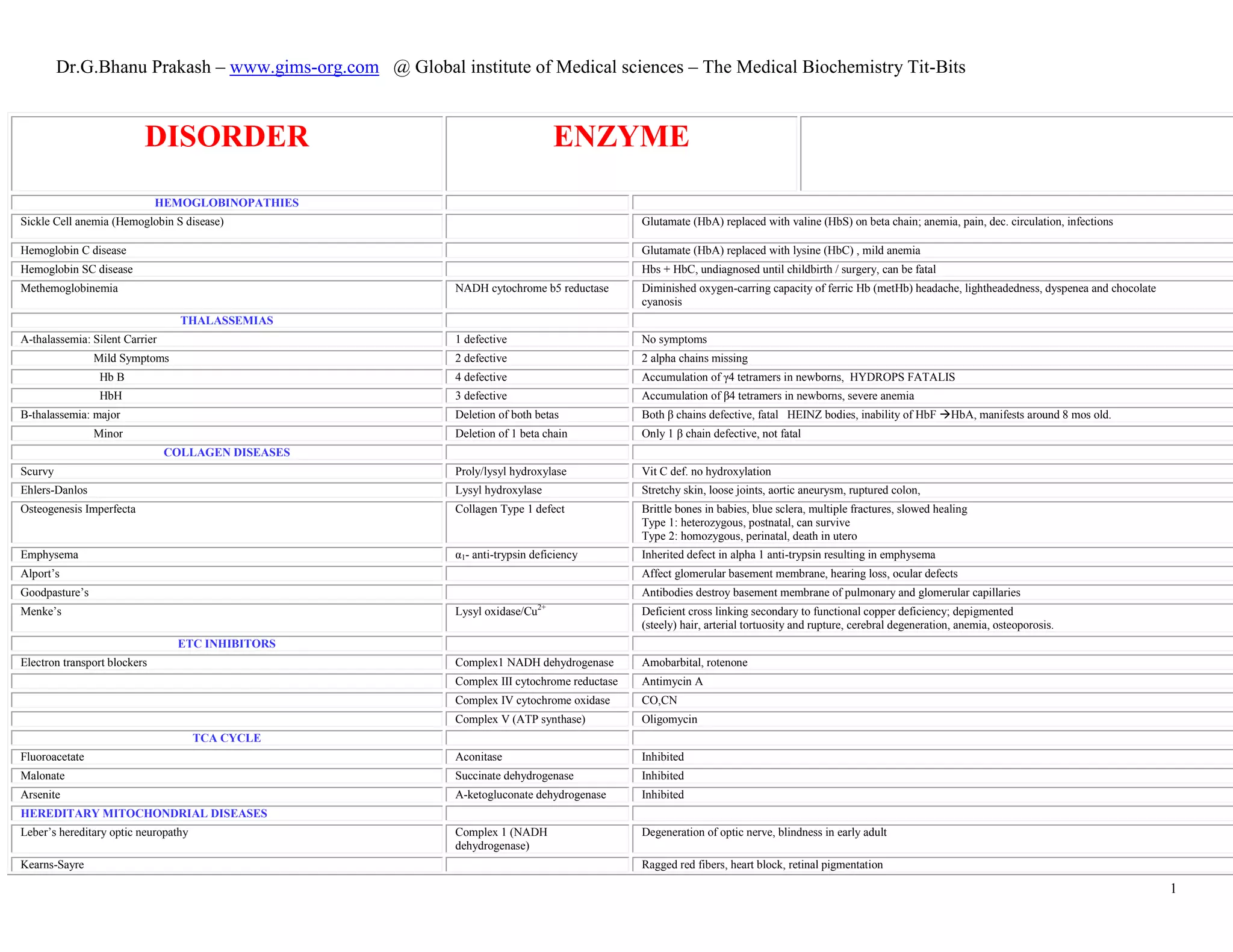
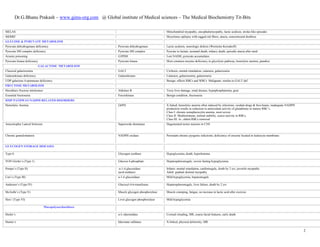
Sanfilippo’s Types A-D Type-Aheparan sulfate
Type BN-
acetylglucosaminidase
Type CN-acetyltransferase
Type D N-acetylglucosamine
Severe nervous system disorders; mental retardation
Scheie’s α-L-iduronidase Like Hurler’s but normal life span
Sly’s β-glucronidase Hepatosplenomegaly, physical deformity
Synthesis of glycoproteins
I-cell disease Lysosomal hydrolytic enzymes Deficiency in ability to phosphorylated mannose residuesof potential lysosomal enzymes;
results in incorrect targeting of glycoproteins; death by 8 yrs; elevated N-linked glycoproteins in urine.
Metabolism of Dietary Lipids
Congenital A-beta-lipoproteinemia Apo B-48 Accum of chylomicrons in enterocytes
Type 1 hyperlipidemia (familial hyperchylomicronemia) Apo C-II, Capillary lipoprotein
lipase
Accumulation of chylomicrons in plasma; high plasma TAGs and eruptive xanthomas (TAG deposits in skin) and
pancreatitis.
Type II hyperlipidemia Genetic defect in synthesis, processing or functioning of LDL receptor; elevated LDL levels
Familial hypercholesterolemia
Type III hyperlipidemia (familial dysbetalipoproteinemia) Apolipoprotein E Accumulation of chylomicron remnants in plasma
Type IV hyperlipidemia Inc. VLDL due to obesity, alcohol, diabeties
Type V hyperlilidemia Inc. chylomicrons, TAGs, VLDL, pancreatitis
Wolman disease Cannot hydrolyze lysosomal cholesteryl esters
Familial LCAT deficiency Complete absence of LCAT, low HDL
Fish Eye Disease Partial LCAT absense
Zellweger syndrome Defective peroxisomal biogenesis, accumulated VLCFAs in blood
X-linked Leukodystrophy Defective peroxisomal activation of VLCFAs, destroyed myelin
Mobilization of Stored Fats & [FA]
Carnitine deficiency inability to use long chain FA as fuel, causes: congenital, liver disease
CAT-1 def Liver cannot synthesize glucose during fast, hypoglycemia, coma, death
CAT-2 def Cardiomyopathy, muscle weakness following exercise
Medium chain Fattyacyl CoA dehydrogenase deficiency Medium chain fatty acyl CoA Decreased FA oxidation, severe hypoglycemia; cause of 10% SIDS cases, Reyes syndrome; treat with
high carb diet
Paroxysomal Nocturnal Hemoglobinuria GPI synthase In hematopoietic cells
Refsum disease Fatty acid alpha-hydroxylase AR, increased phytanic acid, neurologic symptoms](https://image.slidesharecdn.com/biochemistrychart-dr-151101030411-lva1-app6891/85/Biochemistry-chart-dr-g-bhanu-prakash-3-320.jpg)



This document summarizes many metabolic disorders organized by the affected enzyme, pathway, or type of disorder. Some key disorders mentioned include: - Sickle cell anemia caused by a mutation replacing glutamate with valine on the beta globin chain. - Various thalassemias caused by defects in alpha or beta globin chain production leading to anemia. - Phenylketonuria caused by a defect in phenylalanine hydroxylase leading to intellectual disability if untreated. - Glycogen storage diseases caused by defects in glycogen breakdown enzymes leading to hypoglycemia. - Urea cycle disorders caused by enzyme defects preventing ammonia detoxification leading to hyperammone
























![VITAMIN K, [MEDICINAL CHEMISTRY] BY P.RAVISANKAR,STRUCTURES OF VITAMIN K1 AND...](https://cdn.slidesharecdn.com/ss_thumbnails/phm10vitk-130615194322-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)


















































































