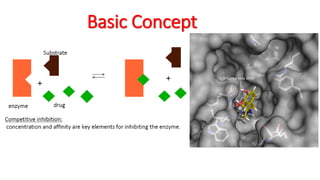
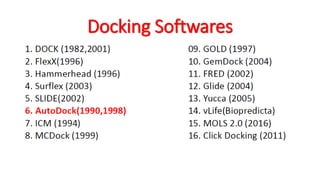
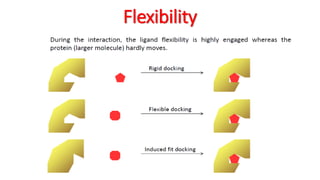
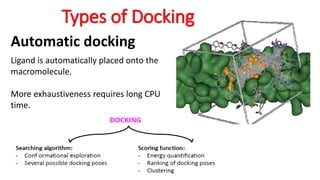
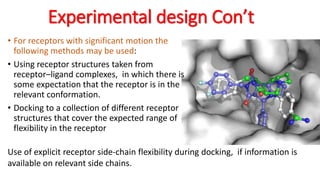
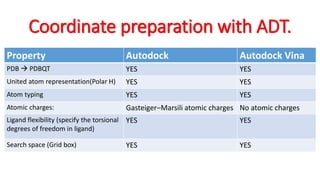
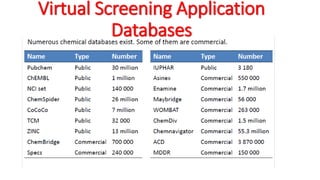
The document reviews the Autodock suite, a set of free open-source tools for computational protein-ligand docking and virtual drug screening, detailing its components such as Autodock, Autodock Vina, Raccoon2, and Autoligand. It discusses the methodology, limitations, and applications in drug design, emphasizing the importance of accurate receptor and ligand preparation for effective docking results. The suite is particularly valuable in predicting binding conformations and free energies, although it faces challenges with large ligands and conformational flexibility of targets.



















































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)






