Downloaded 238 times







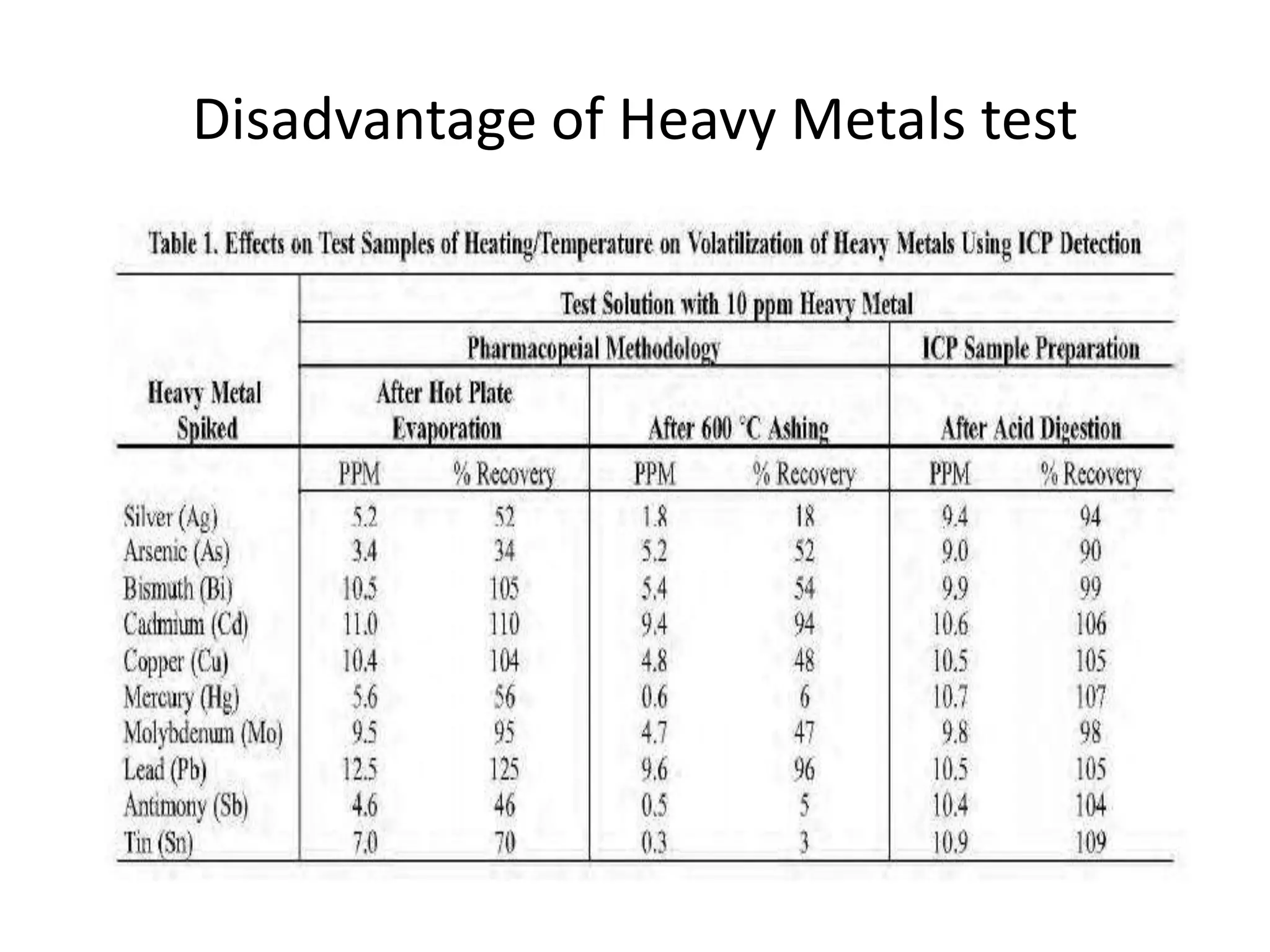


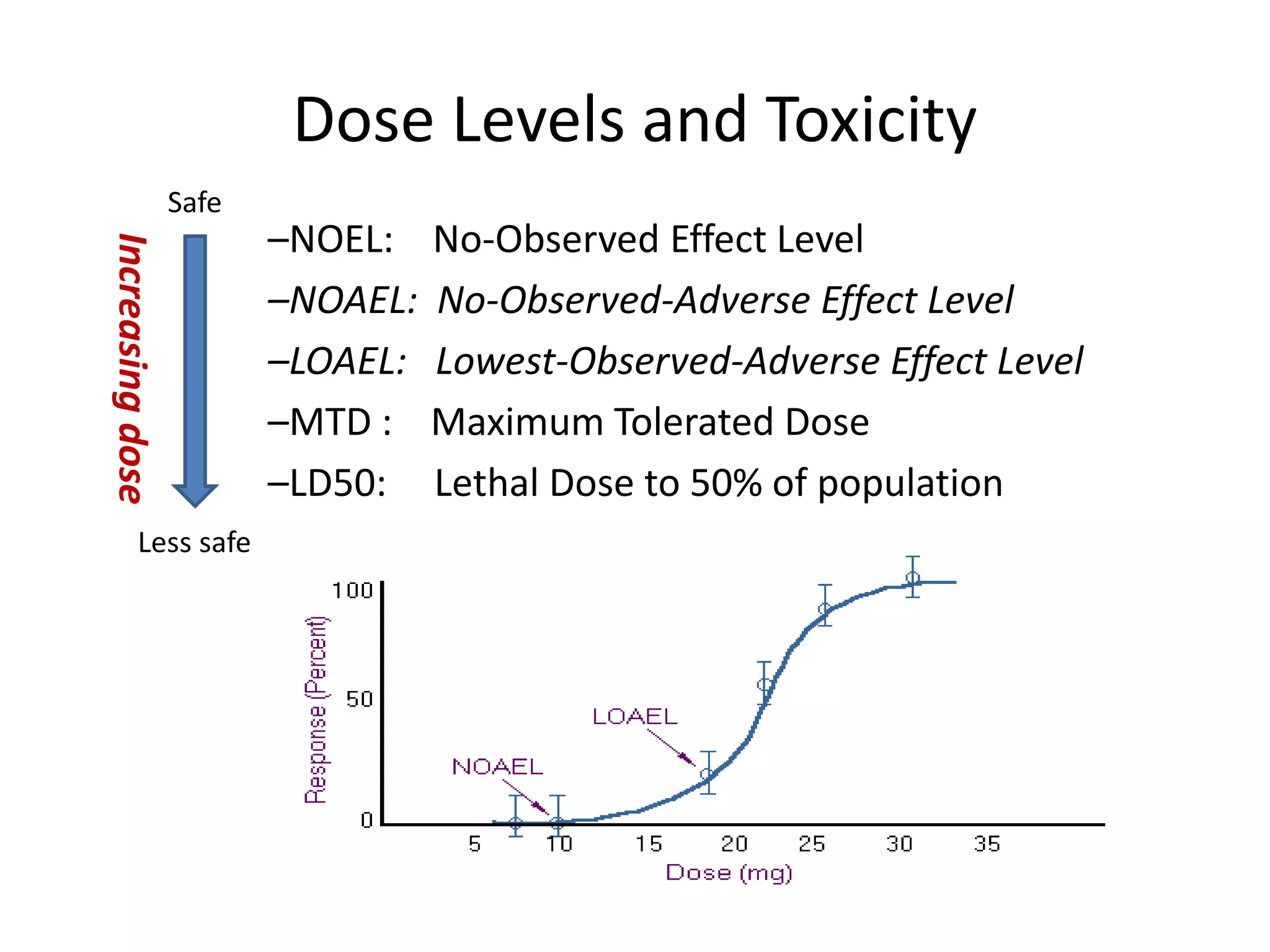








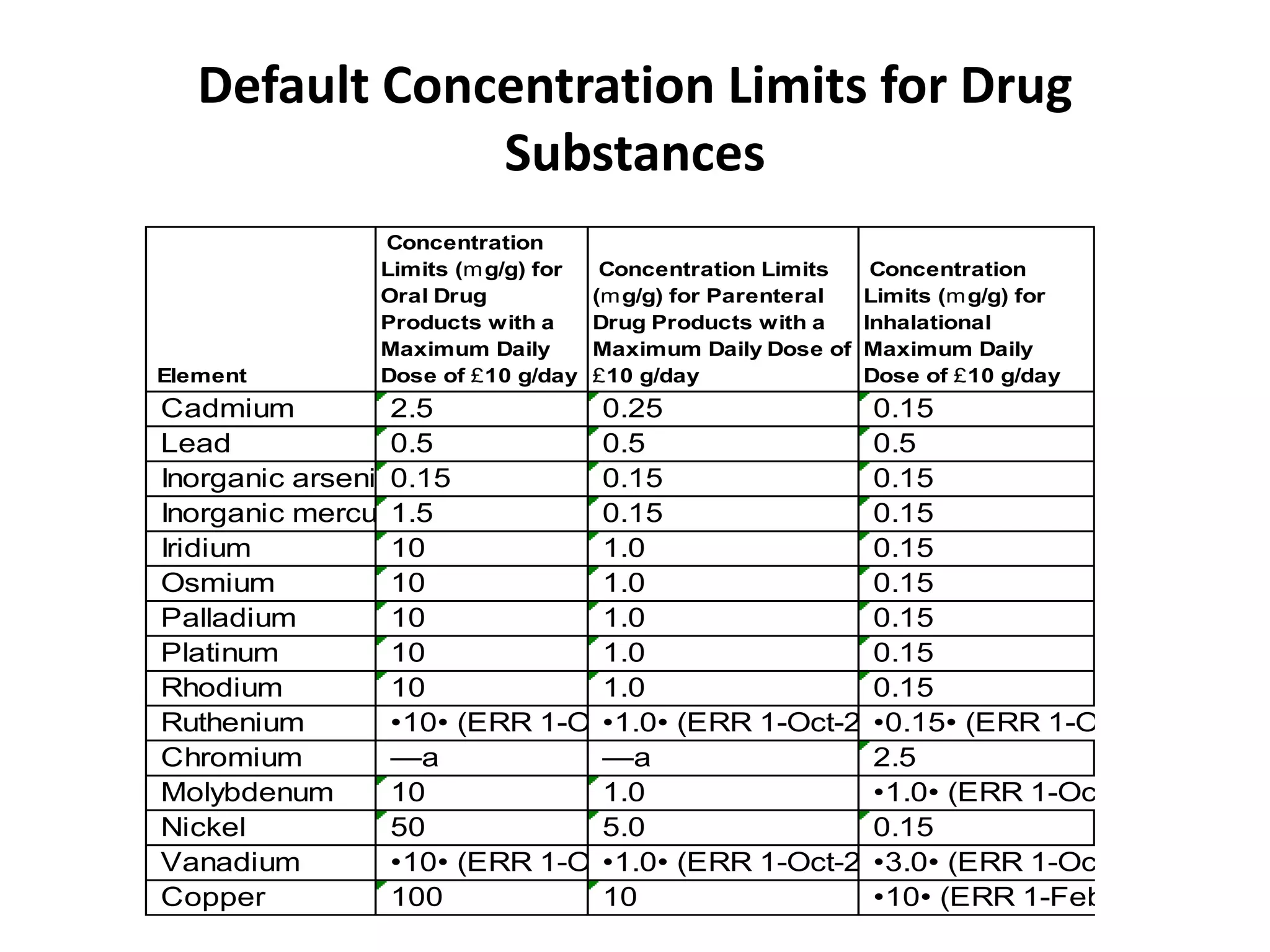

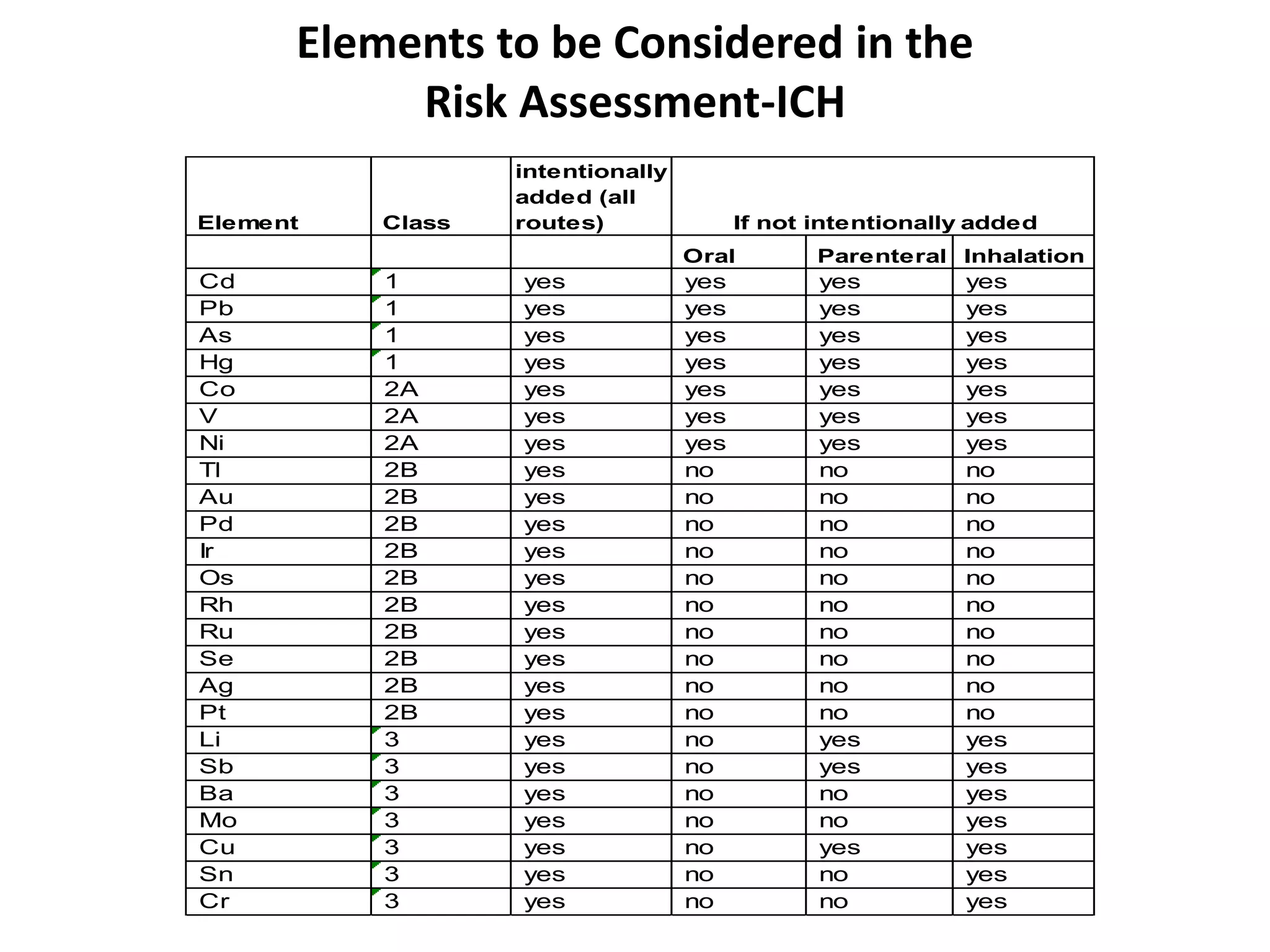
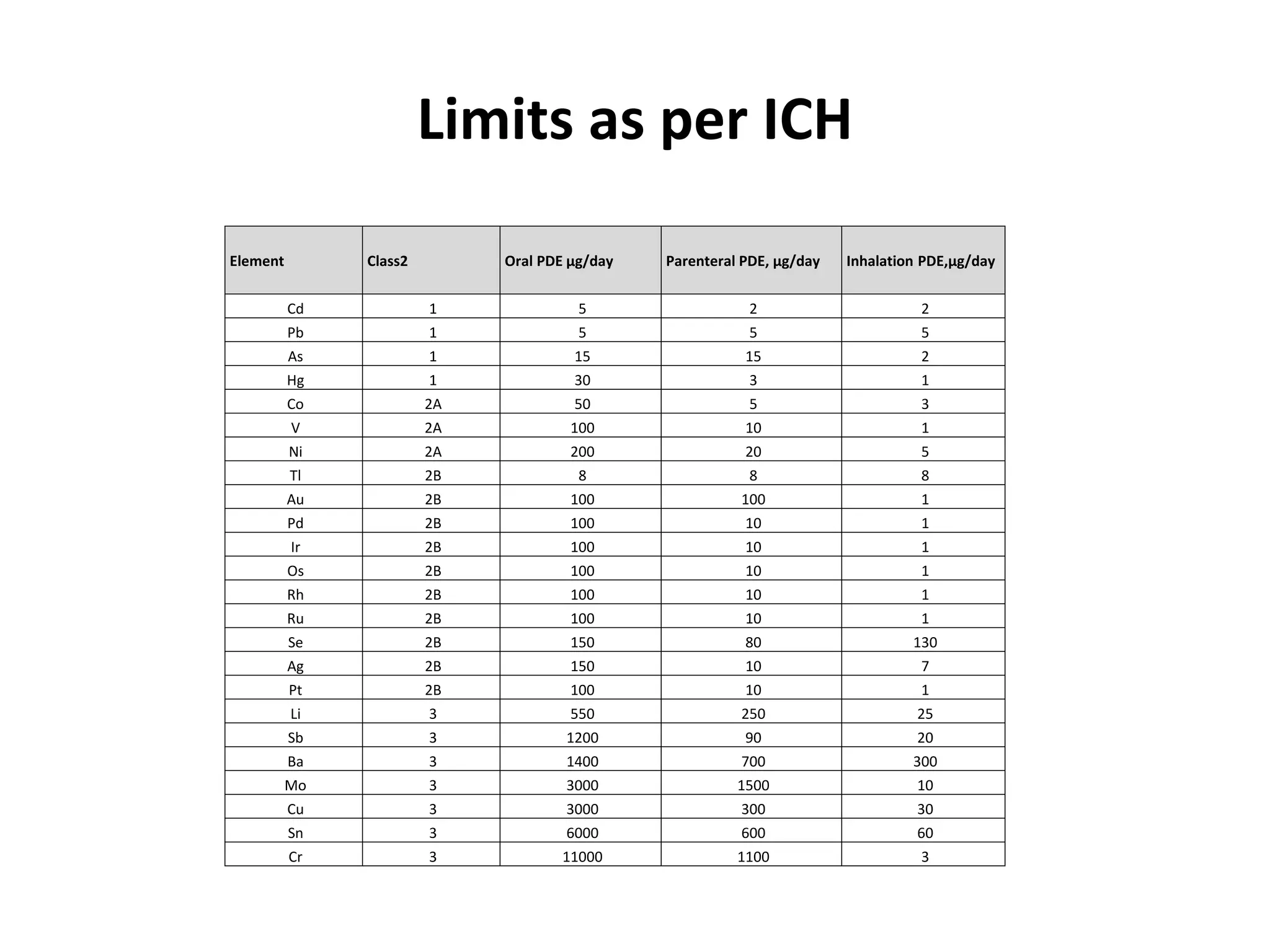

































The document discusses guidelines for controlling elemental impurities in active pharmaceutical ingredients (APIs) according to new regulatory requirements. It provides an overview of: 1) Background guidelines from various regulatory agencies on limiting elemental impurities. 2) Reasons for the new requirements to replace heavy metal testing, including difficulties with reproducibility and safety of current methods. 3) Classification of elemental impurities based on toxicity and permissible intake limits set by the ICH. It also outlines procedures for method development, validation, and implementation of elemental impurity testing and control as defined in USP general chapters 232 and 233.

























![Basics impurity profiling and degradent characterization[134]](https://cdn.slidesharecdn.com/ss_thumbnails/basicsimpurityprofilinganddegradentcharacterization134-191014164210-thumbnail.jpg?width=640&height=640&fit=bounds)











































































![Apporach to lung biopsy [Auto-saved].pptx latest](https://cdn.slidesharecdn.com/ss_thumbnails/apporachtolungbiopsyauto-saved-251211225655-93258539-thumbnail.jpg?width=640&height=640&fit=bounds)





