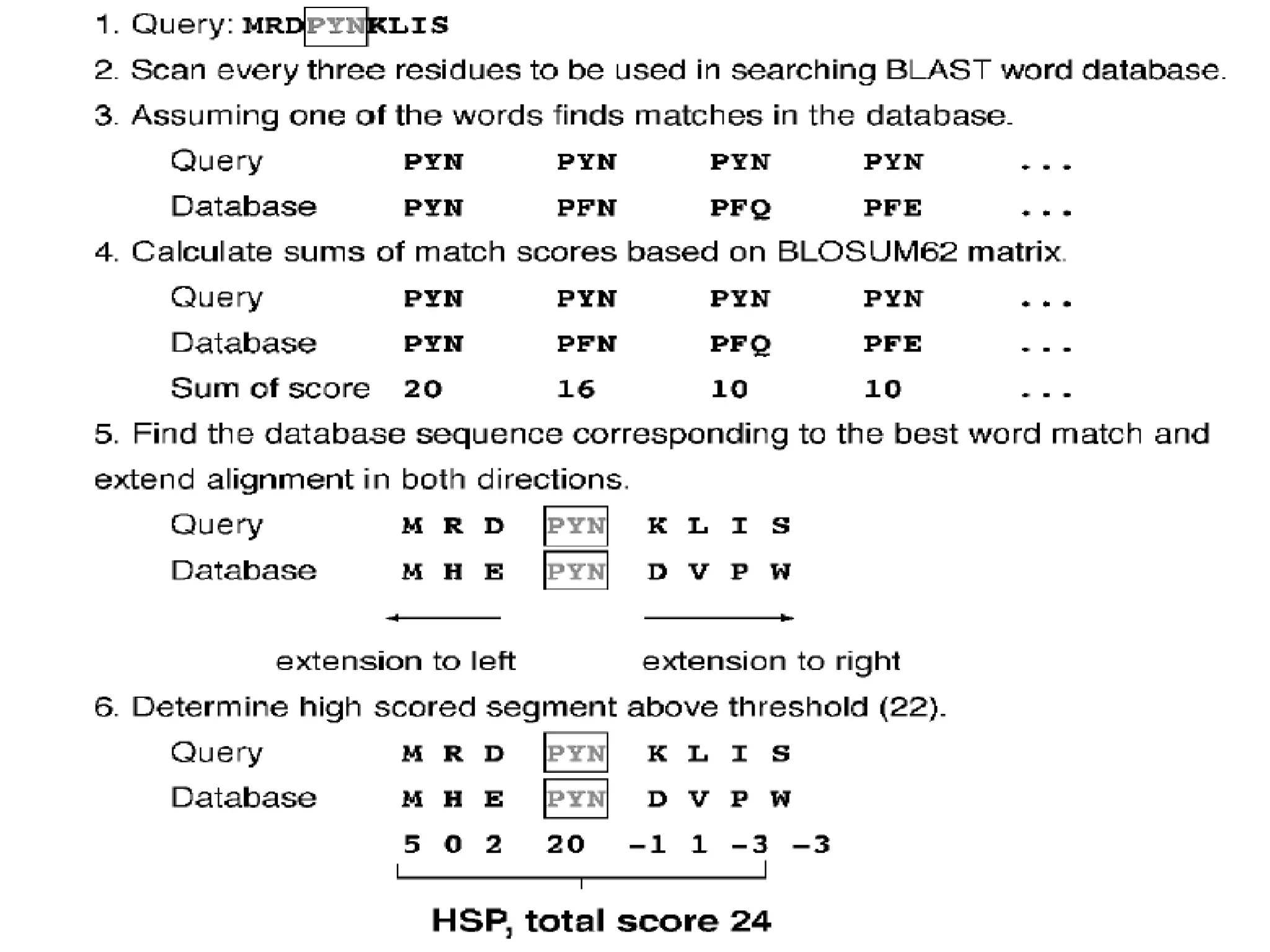
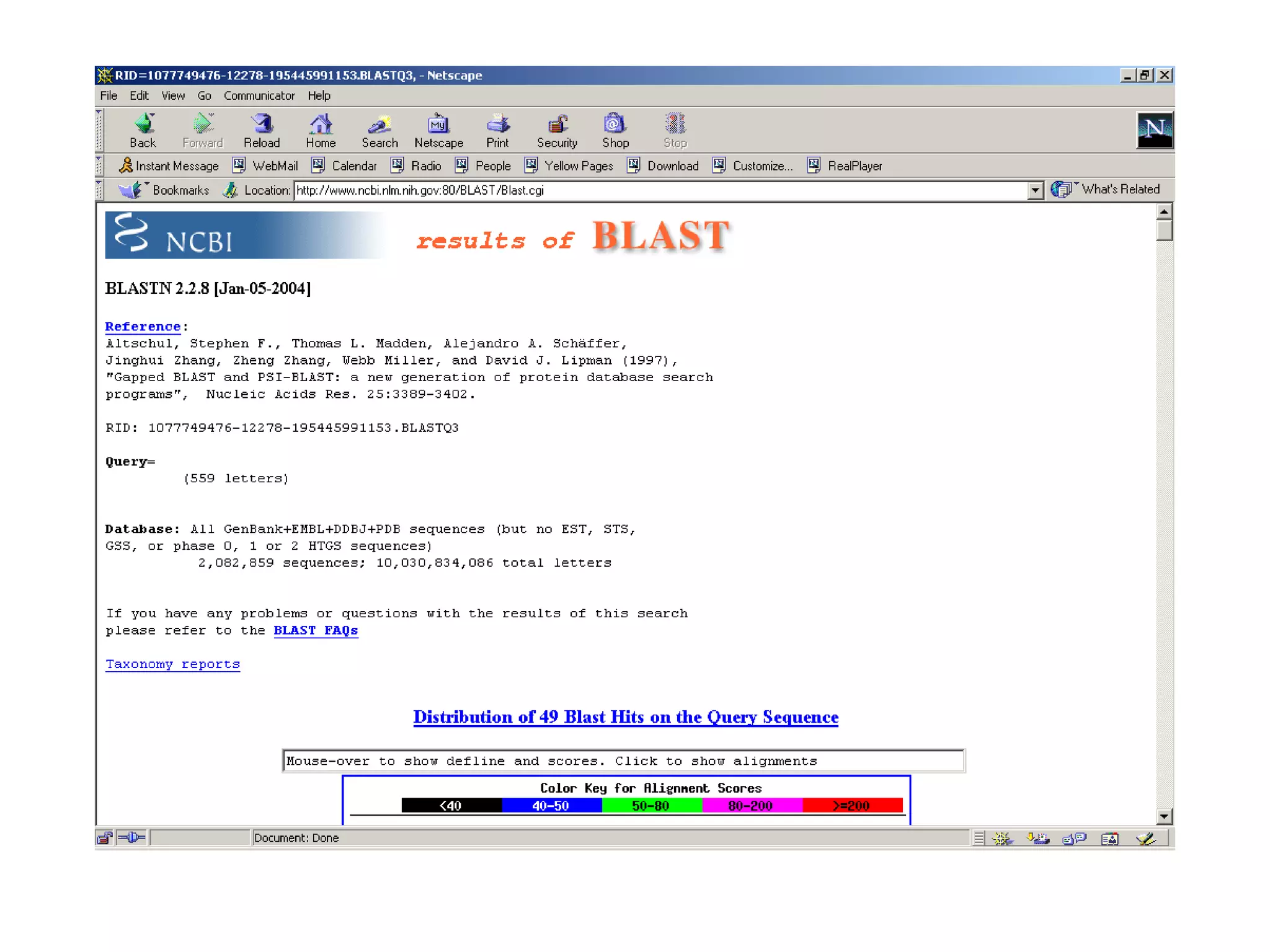
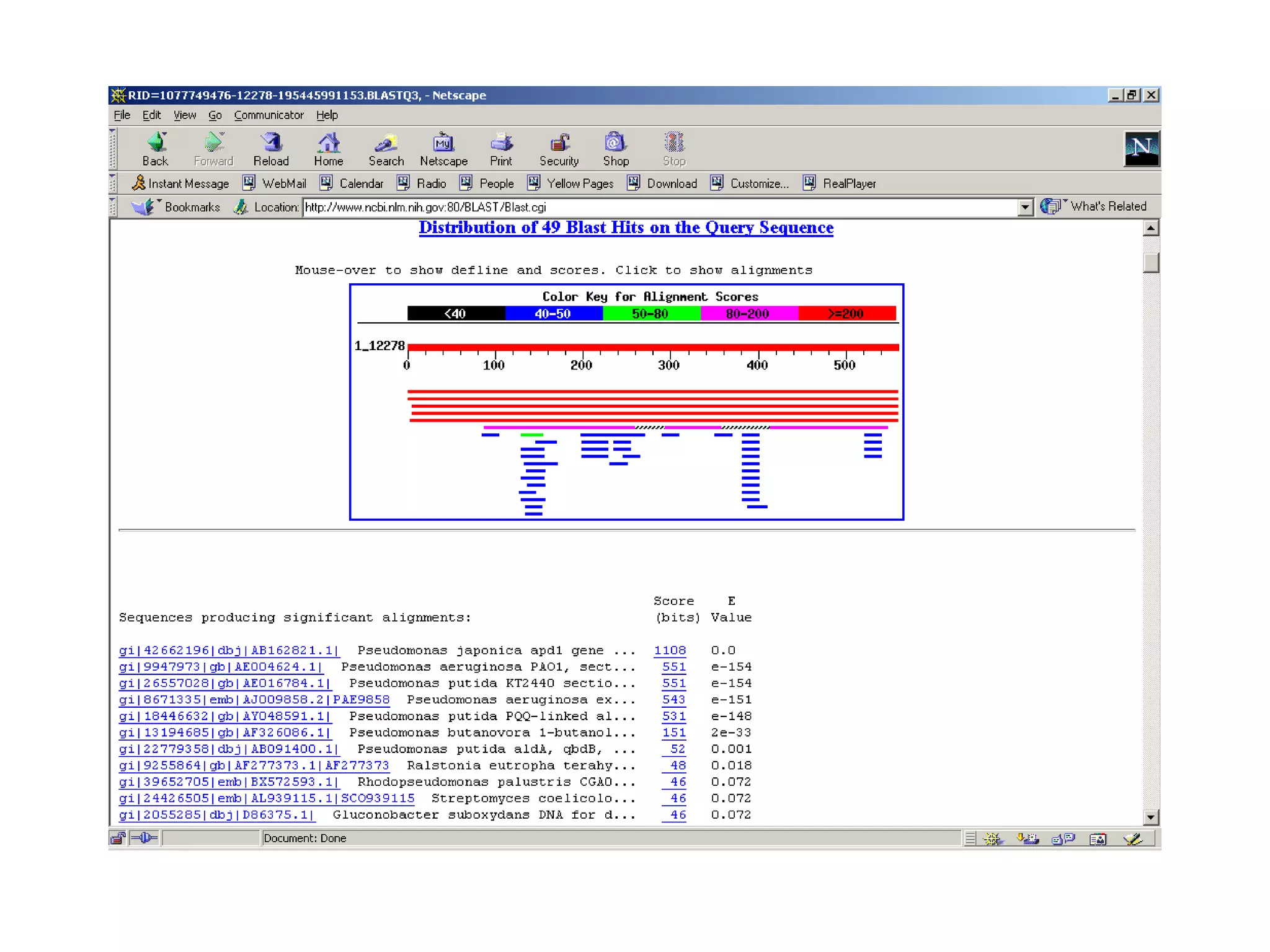
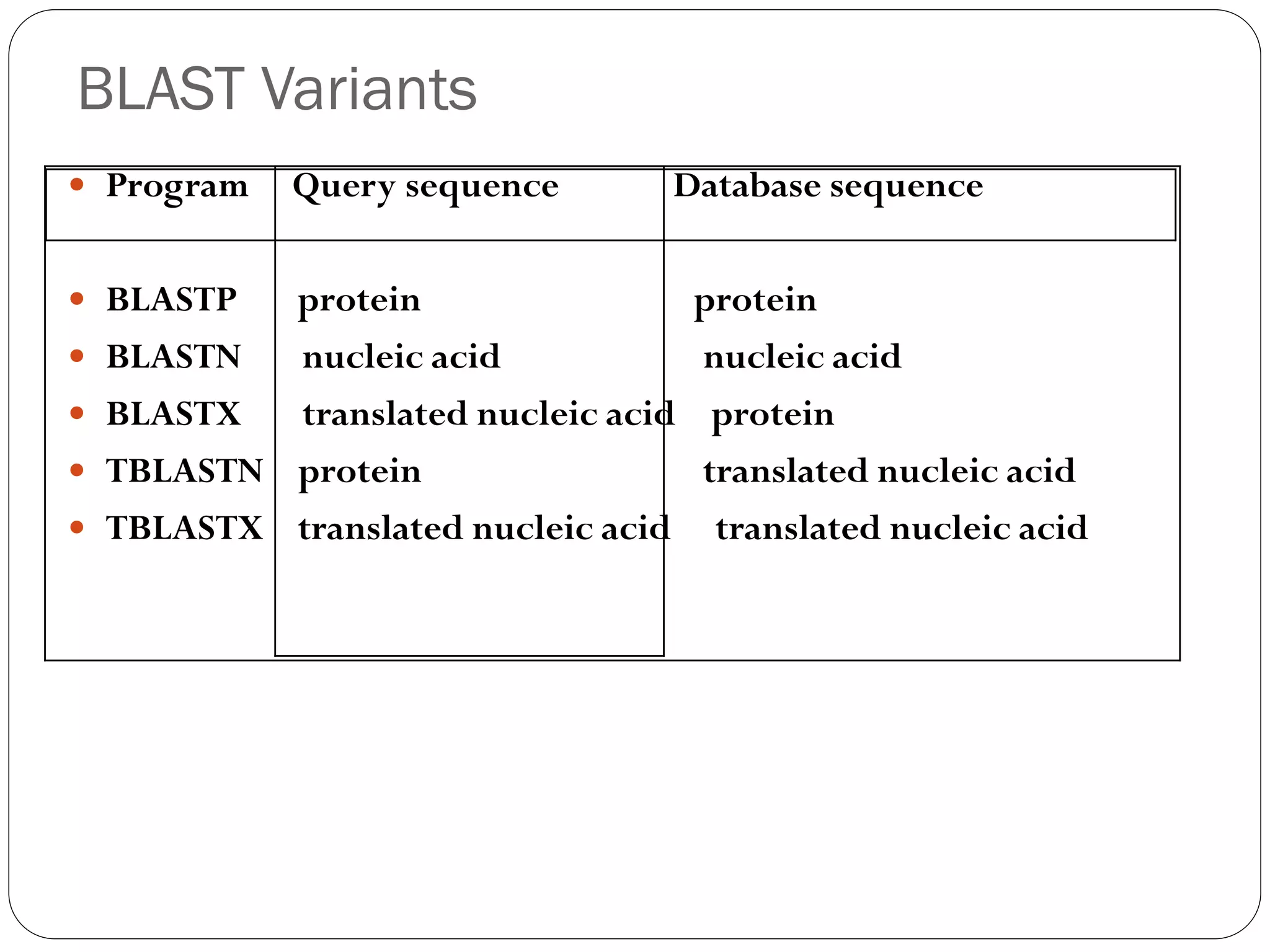
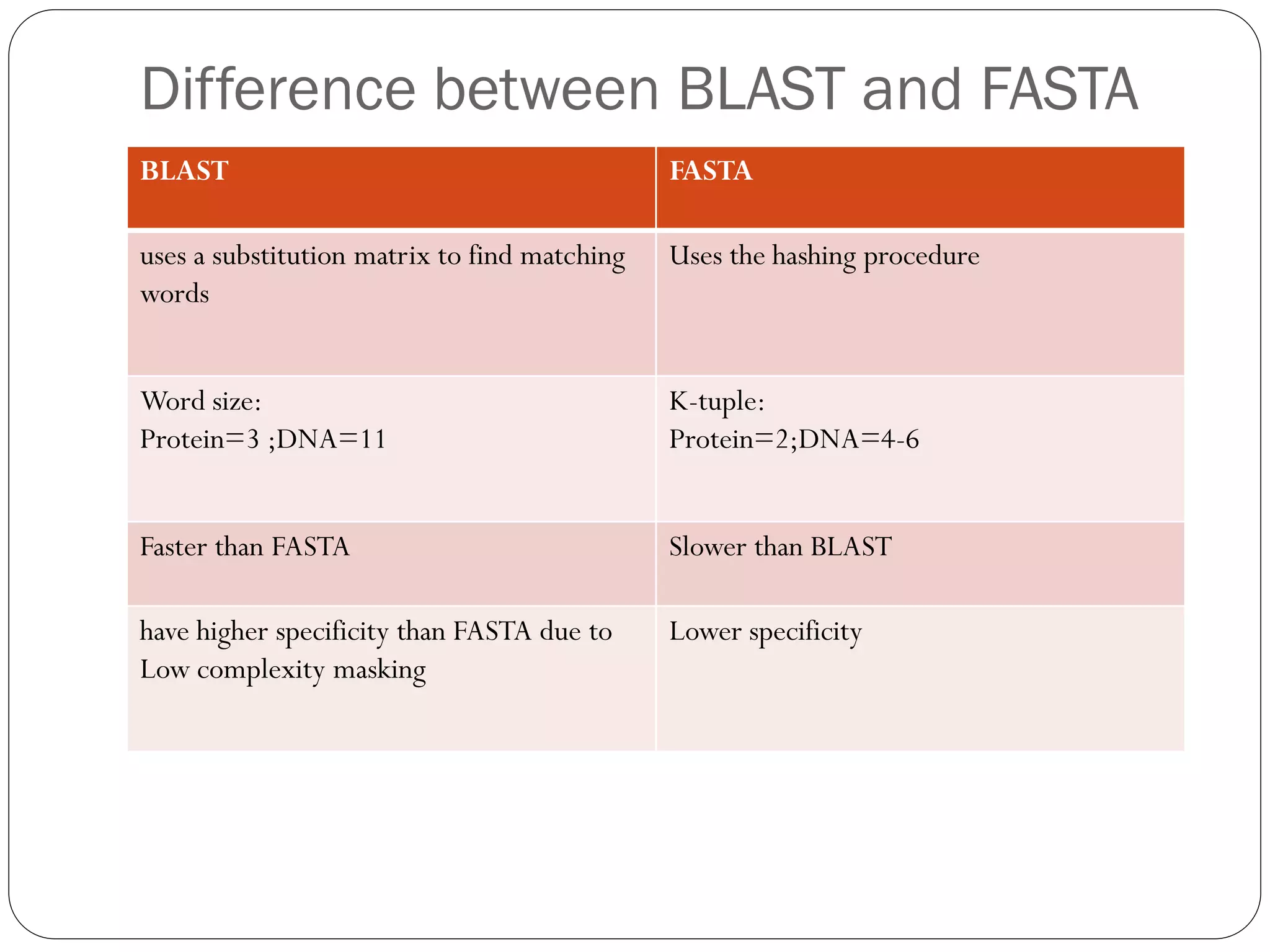
BLAST is a program that compares a query DNA or protein sequence against a database to find sequences that resemble the query above a certain threshold. It works by breaking the query into short words and searching the database for those words, then extending any matches. The output includes alignments of high-scoring segment pairs and statistical measures like E-values and bit scores to indicate match significance. BLAST is faster than FASTA and more specific due to low complexity filtering.













![Databases available on BLAST Web server
Database Description
A. Peptide sequence databases
1.
nr-translations of GenBank DNA sequences with redundancies removed,
PDB,
SwissProt, PIR, and PRF
2.
month -new or revised entries or updates to nr in the previous 30 days
3.
Swissprot- latest release of the SwissProt protein sequence databasea
4.
Drosophila genome -provided by Celera and Berkeley Drosophila genome
project
5.
yeast -yeast (Saccharomyces cerevisiae) genomic sequences
6.
E. Coli- E. coli genomic sequences
7.
pdb -sequences of proteins of known three-dimensional structure from the
Brookhaven Protein Data Bank
8.
yeast -yeast (S. cerevisiae) protein sequences
9.
E. coli- E. coli genomic coding sequence translations
10. kabat [kabatpro] -Kabat’s database of sequences of immunological interest
11. Alu- translations of select Alu repeats from REPBASE, a database of sequence
repeats](https://image.slidesharecdn.com/blast-131109005515-phpapp01/75/Blast-bioinformatics-18-2048.jpg)
![ B. Nucleotide sequence databases
1. nr- GenBank, EMBL, DDBJ, and PDB sequences with redundancies
removed (EST, STS, GSS, and HTGS sequences excluded)
2. month -new or revised entries or updates to nr in the previous 30
days
3. dbestb- EST sequences from GenBank, EMBL, and DDBJ with
redundancies removed
4. dbstsb- STS sequences from GenBank, EMBL, and DDBJ with
redundancies removed
5. htgsb- high-throughput genomic sequences
6. kabat [kabatnuc] -Kabat’s database of sequences of immunological
interest
7. vector- vector subset of GenBank
8. mito -database of mitochondrial sequences
9. alu -select Alu repeats from REPBASE, a database of sequence repeats;
suitable for masking Alu repeats from query sequences
10. epd- eukaryotic promoter database
11. gssb -genome survey sequences, includes single-pass genomic
data,exon-trapped sequences, and Alu PCR sequences](https://image.slidesharecdn.com/blast-131109005515-phpapp01/75/Blast-bioinformatics-19-2048.jpg)






































![BLAST [Basic Alignment Local Search Tool]](https://cdn.slidesharecdn.com/ss_thumbnails/blast-120911083837-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)


































![Vibe Coding vs. Spec-Driven Development [Free Meetup]](https://cdn.slidesharecdn.com/ss_thumbnails/vibecodingvsspecdrivendevelopment-251209105622-43f455e7-thumbnail.jpg?width=640&height=640&fit=bounds)

















