






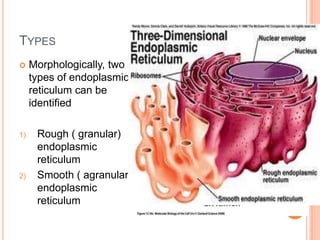


















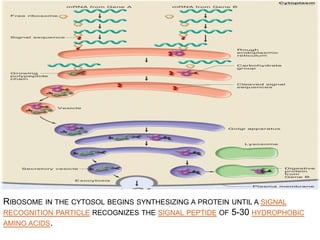
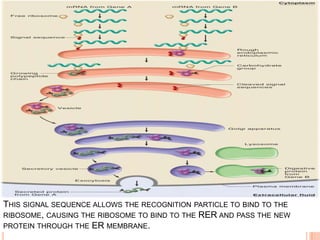
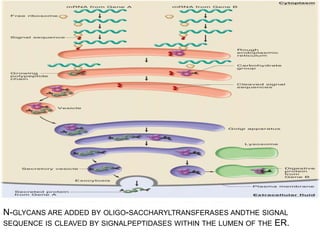
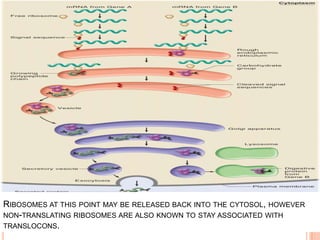


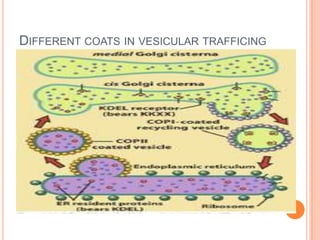


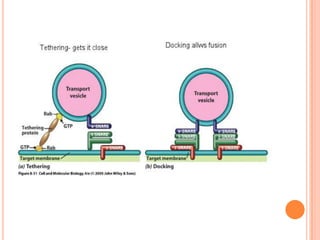
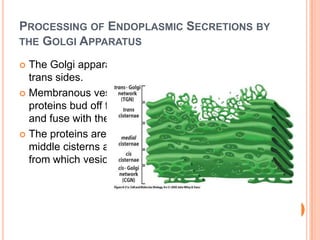


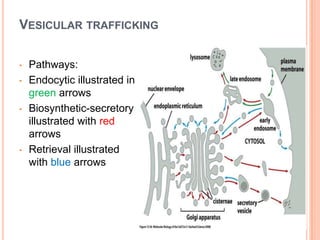
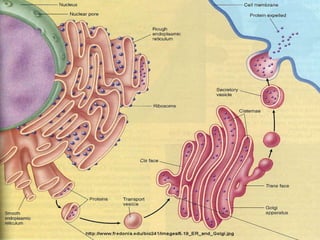







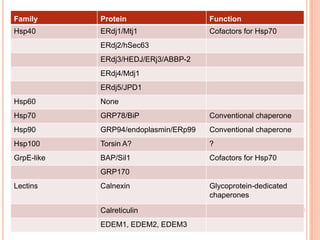






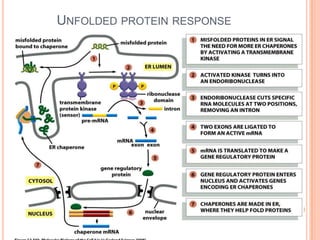






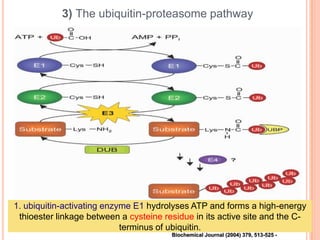
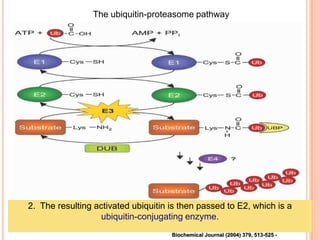
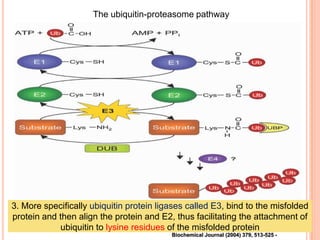
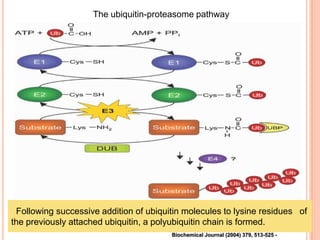
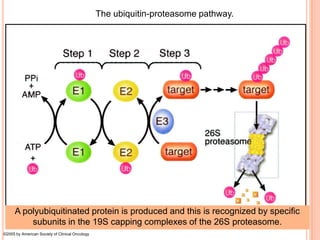






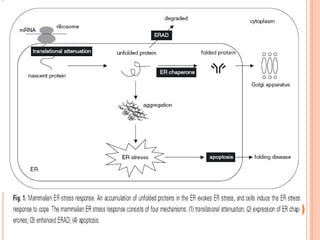



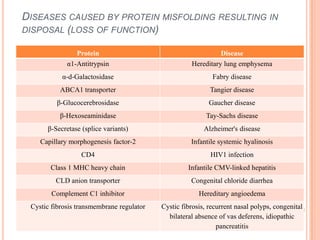
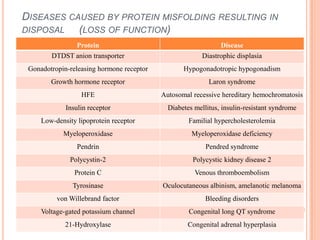


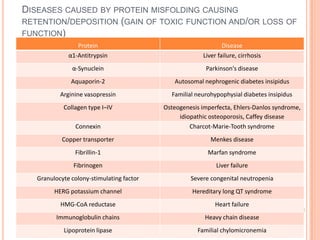
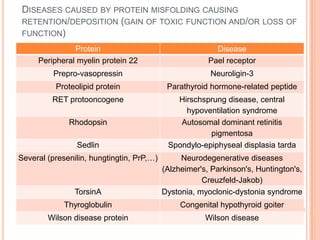


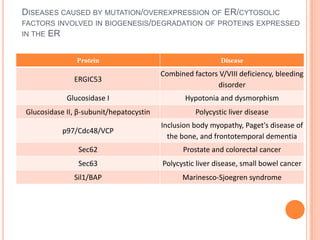



The endoplasmic reticulum (ER) is an organelle found in eukaryotic cells that forms an interconnected network of tubules, vesicles, and cisternae. It has two main types - rough ER with ribosomes and smooth ER without. The rough ER is involved in protein synthesis and modification, while the smooth ER performs functions like lipid synthesis and calcium regulation. Newly synthesized proteins are transported from the ER to the Golgi apparatus in vesicles for further processing and modification before being packaged into secretory vesicles and transported throughout the cell. The ER also plays a key role in protein folding and quality control.















































![Endoplasmic reticulum[1]](https://cdn.slidesharecdn.com/ss_thumbnails/endoplasmicreticulum1-160424155701-thumbnail.jpg?width=640&height=640&fit=bounds)


































