Downloaded 558 times


















![Regulatory Overview
Federal Food, Drug, and Cosmetic Act (the Act)[21 U.S.C
§ 331(a)(2)(B)]
Methods, Facilities, and Controls must comply with
cGMP or the drug product is deemed adulterated!
Need to provide the evidence that review of the
Batch Record including all critical step.
The above confirms that the product meets cGMPs
with the Batch Record documentation.](https://image.slidesharecdn.com/bprreviewandbatchrelease-190906174410/85/BPR-review-and-batch-release-18-320.jpg)













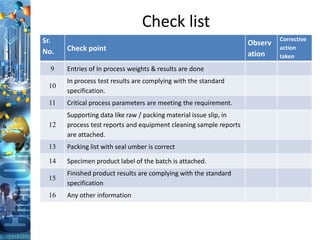
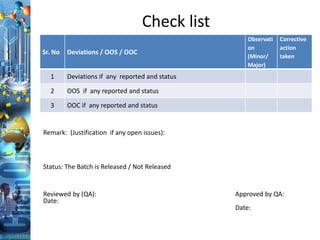
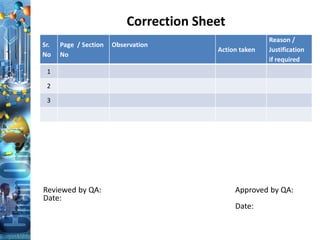








The document discusses batch production record (BPR) review and release. It defines key terms like deviations, critical process parameters, critical quality attributes. It outlines regulatory requirements from ICH Q7, CFR 211, and consequences of non-compliance. The objectives of BPR review are to confirm the batch quality and was produced under control. Records of critical steps must be reviewed and approved by quality before release. Failure to comply with cGMPs can render a drug adulterated under the FDA act.







































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)







![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)







