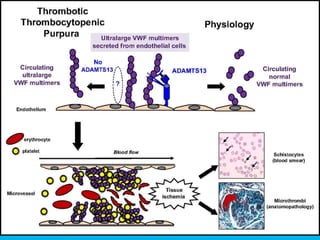
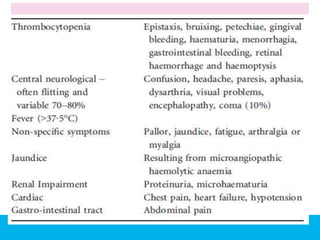
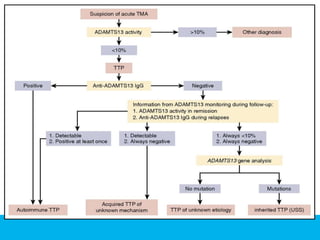
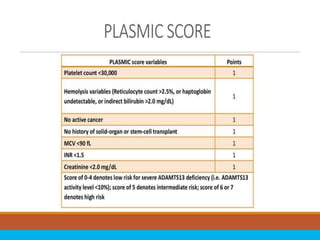
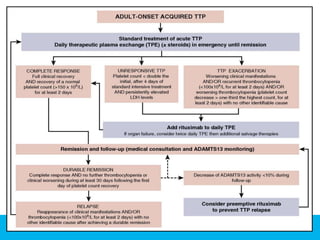
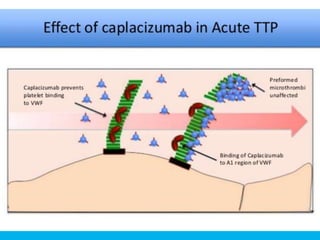
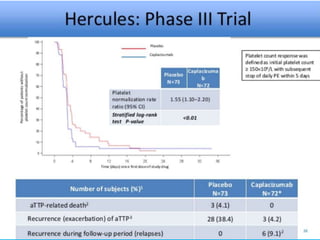
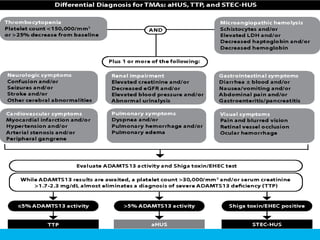
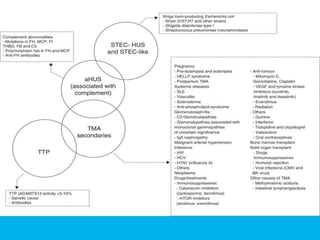
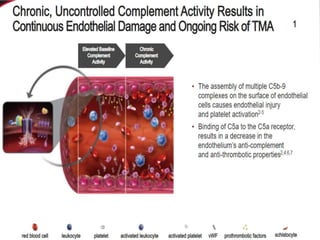
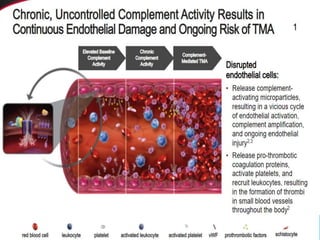
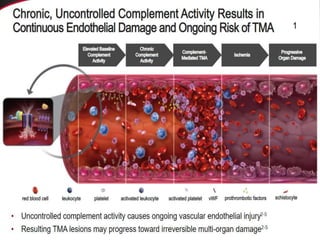
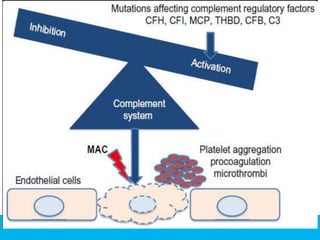
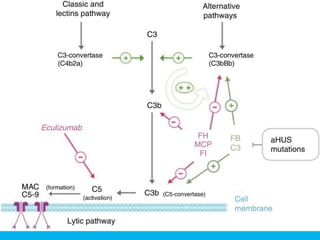
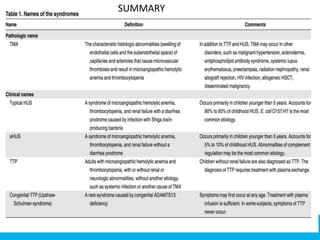
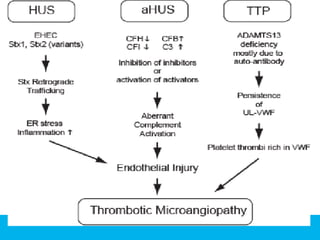
The document discusses thrombotic thrombocytopenic purpura (TTP) and atypical hemolytic uremic syndrome (HUS) as distinct diseases, highlighting epidemiology, diagnosis, and types of TTP. It emphasizes the mortality rates associated with TTP and the classic symptoms such as microangiopathic hemolytic anemia and thrombocytopenia. Finally, it advocates for the discontinuation of the combined term 'TTP/HUS' in medical terminology.