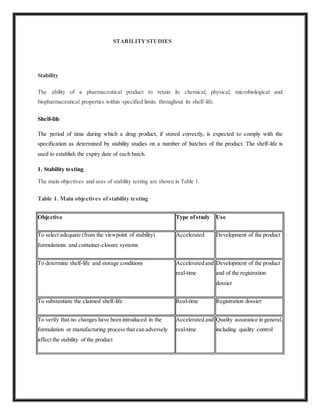
This document discusses stability studies that are conducted on pharmaceutical products to ensure they maintain their chemical, physical, and other properties throughout their shelf life. It addresses the objectives of stability testing including selecting formulations and packaging, determining shelf life and storage conditions, and quality assurance. It also discusses the design of stability studies, including test samples, test conditions for accelerated and real-time studies, and intended market climates. The goal of stability testing is to confirm that a pharmaceutical product is of acceptable quality for its labeled shelf life.