Downloaded 33 times
























![EDSS PLUS
EDSS +
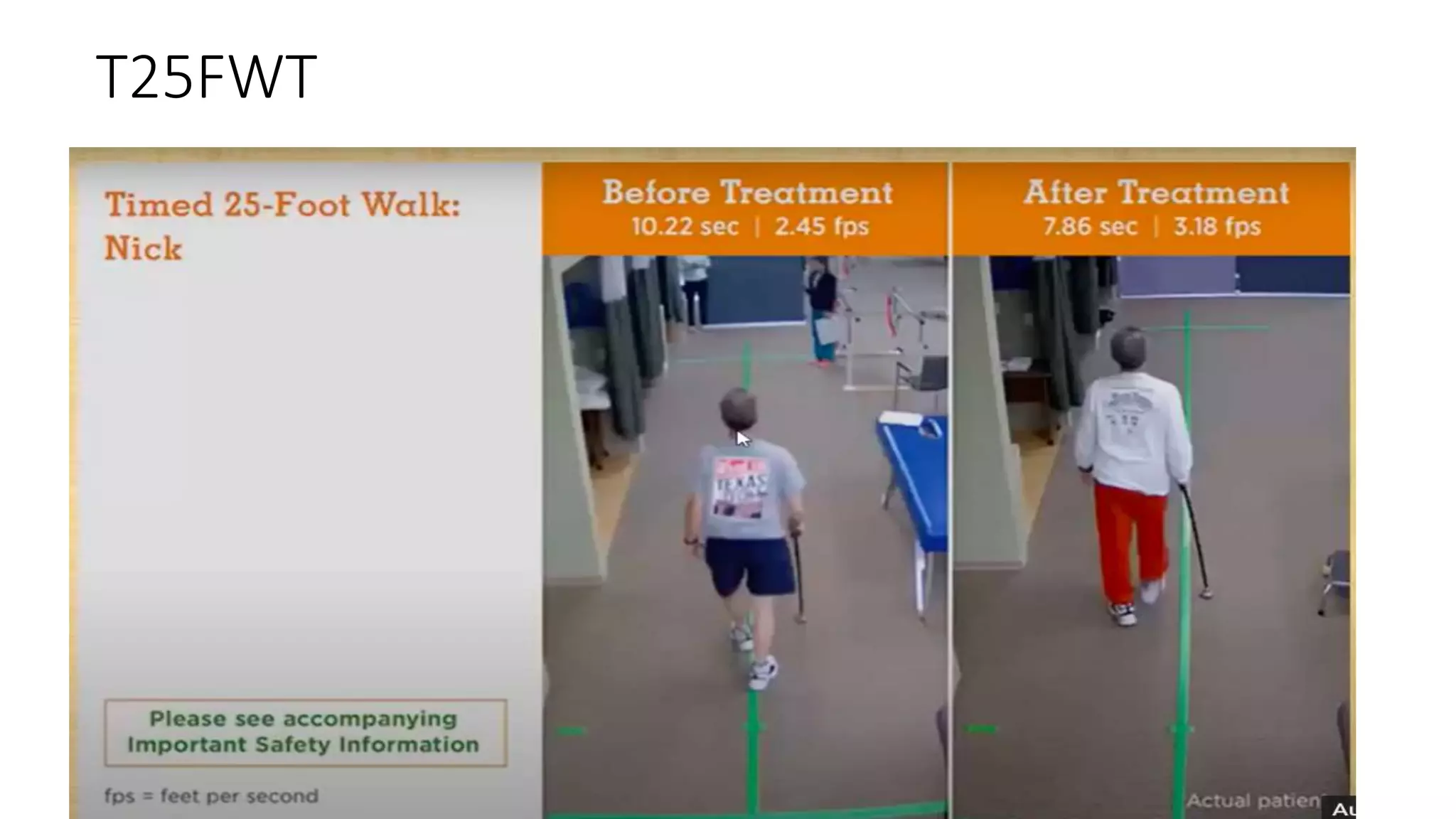
-(timed 25-foot walk)
-(9-hole peg test): upper extremity function
[a test of manual dexterity in which the time to place nine pegs into a
3 x 3 array is recorded]](https://image.slidesharecdn.com/progressivems-220124213154/75/Progressive-ms-32-2048.jpg)
![Multiple Sclerosis Functional
Composite (MSFC)
-(timed 25-foot walk)
-(9-peg hole test): upper extremity function
[a test of manual dexterity in which the time to place nine pegs into a
3 x 3 array is recorded]
-Cognitive function (Paced Auditory serial addition test)](https://image.slidesharecdn.com/progressivems-220124213154/75/Progressive-ms-33-2048.jpg)






























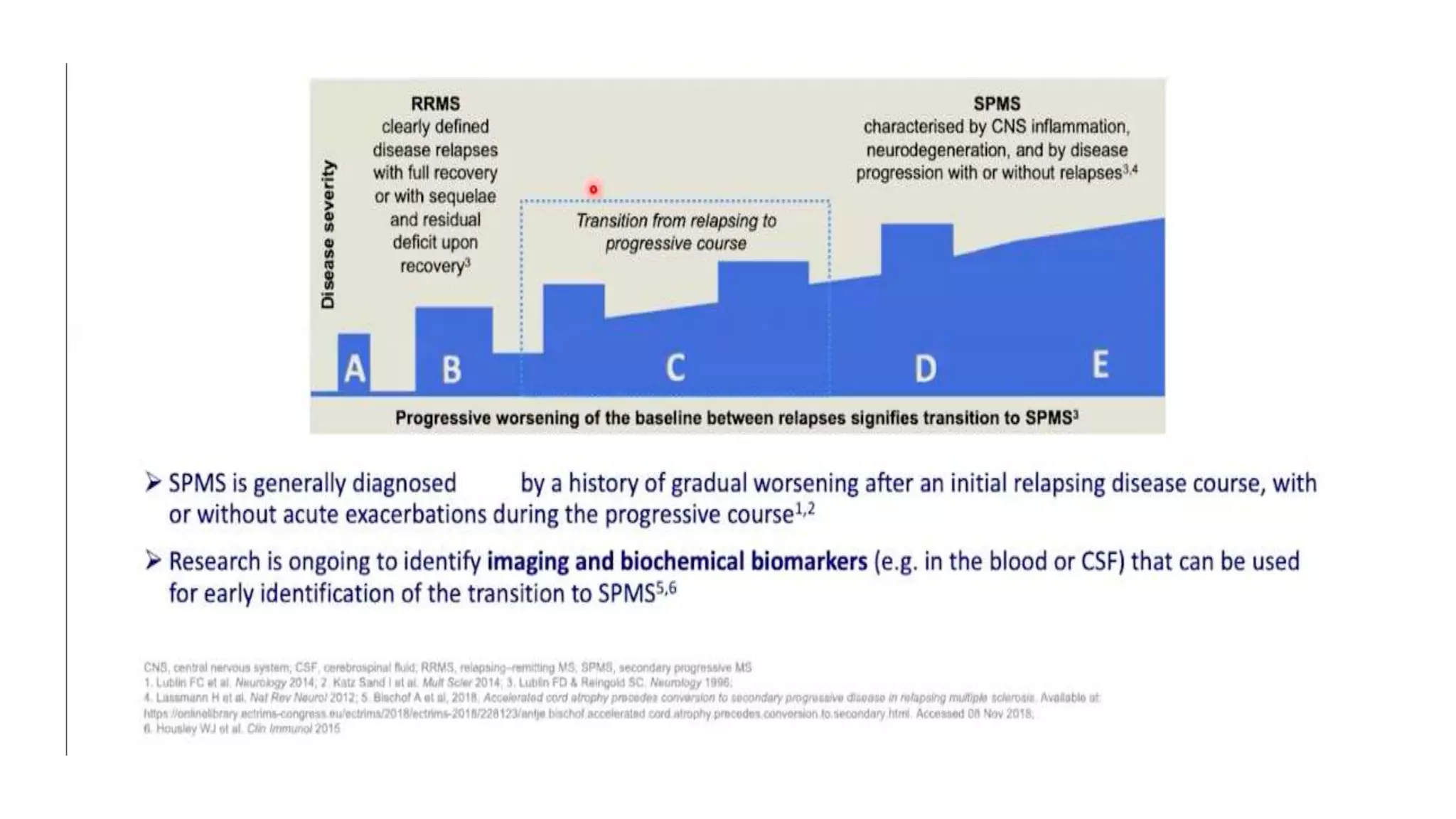
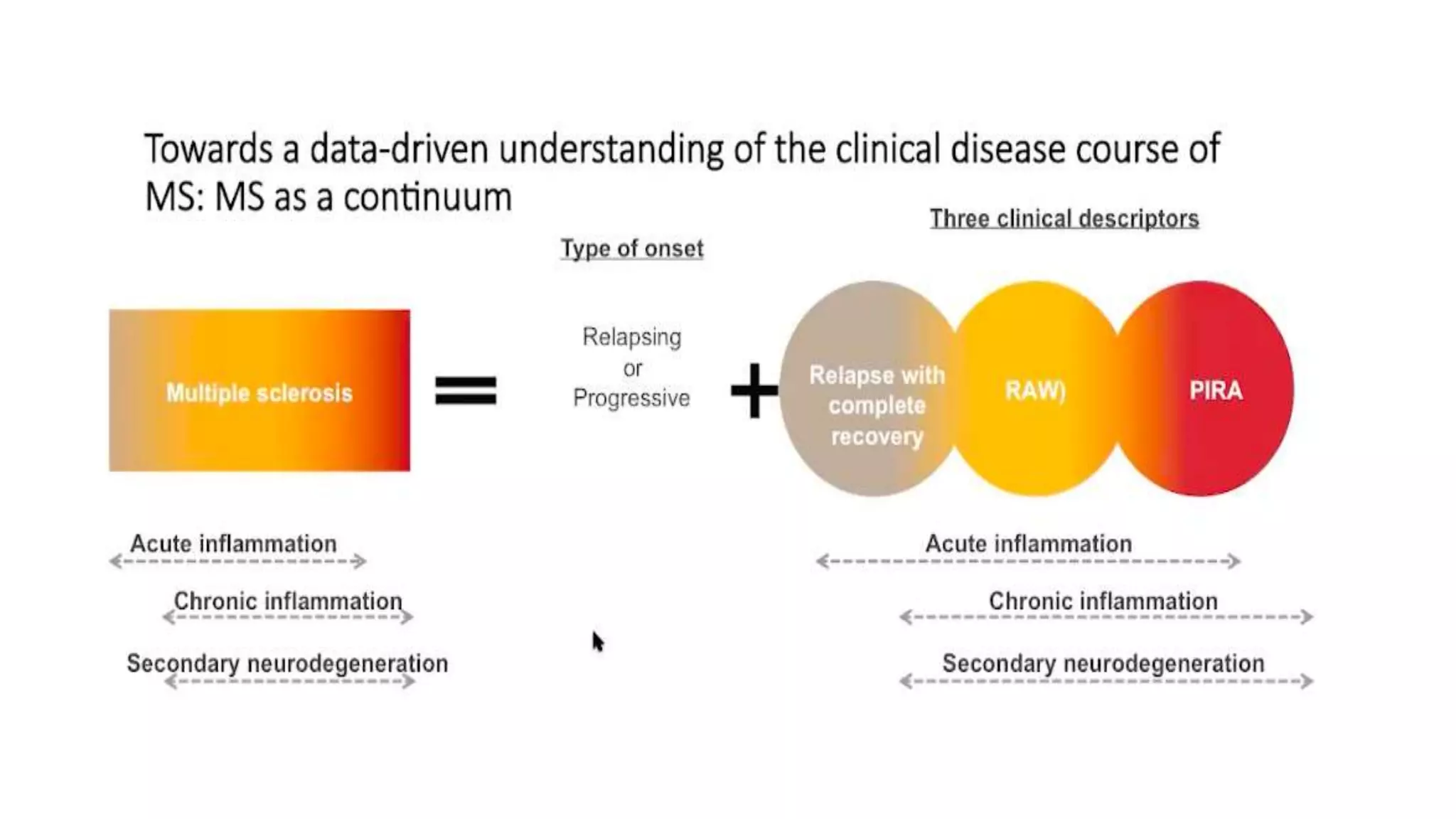
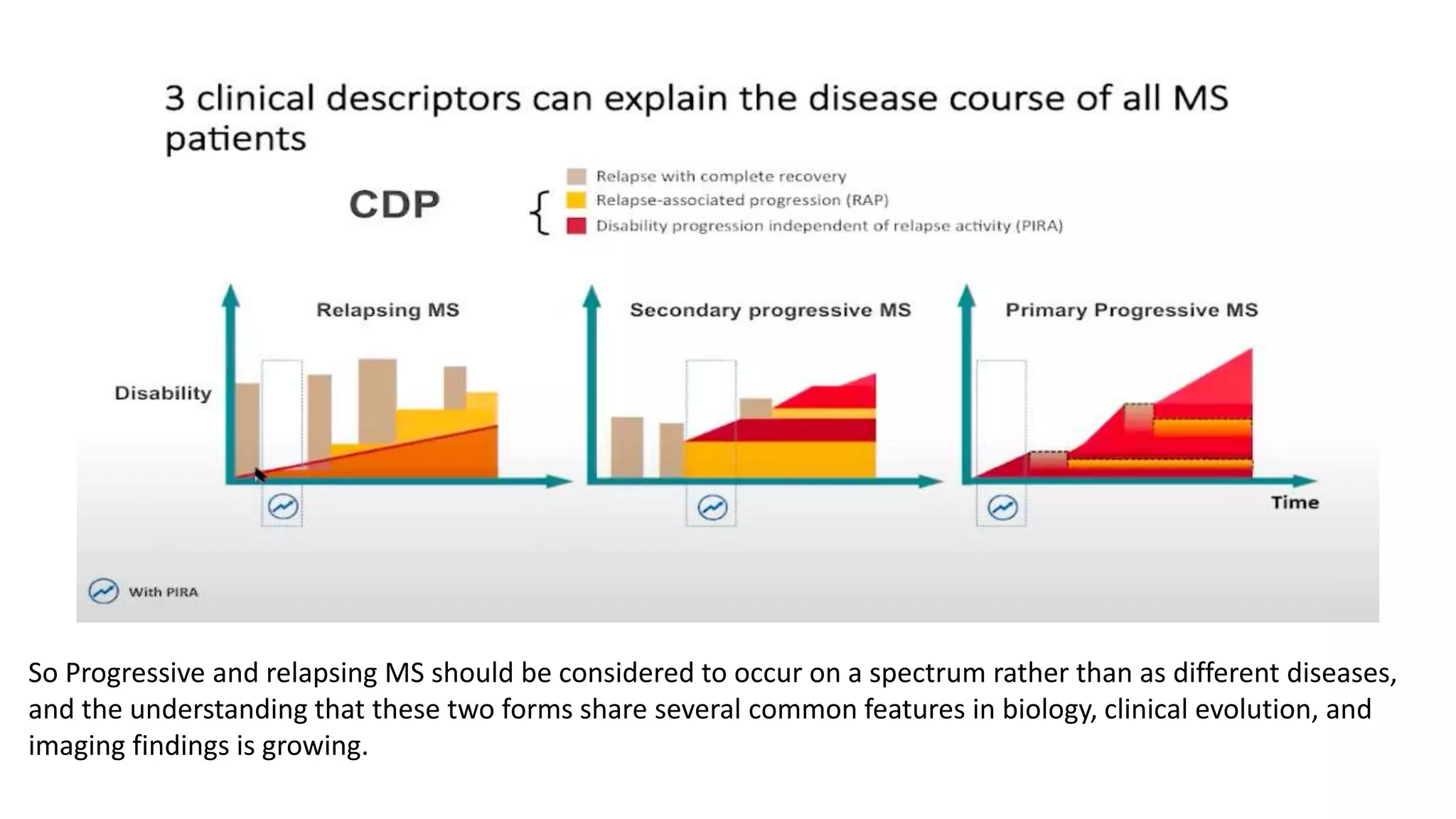
Progressive multiple sclerosis (MS) can be primary progressive or secondary progressive and occurs on a spectrum with relapsing MS. Treatment approaches for progressive MS include immunomodulation, B-cell therapies like ocrelizumab which is approved for primary progressive MS, and neuroprotective agents. Monitoring for progression may involve markers like neurofilament light chain in serum and cerebrospinal fluid as well as optical coherence tomography and spinal cord MRI measures. Management of progressive MS also focuses on controlling medical comorbidities.









































































