Downloaded 1,508 times


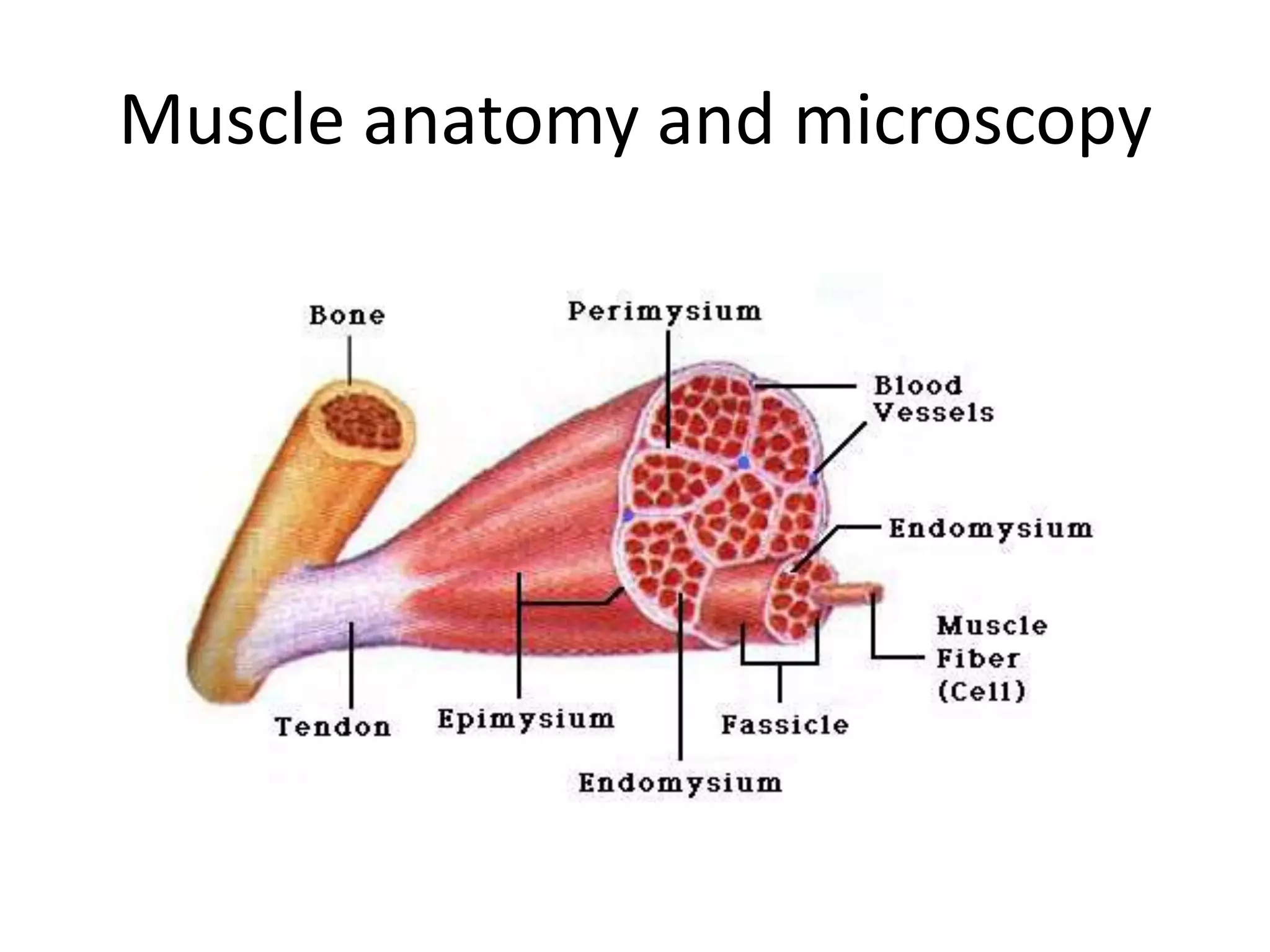
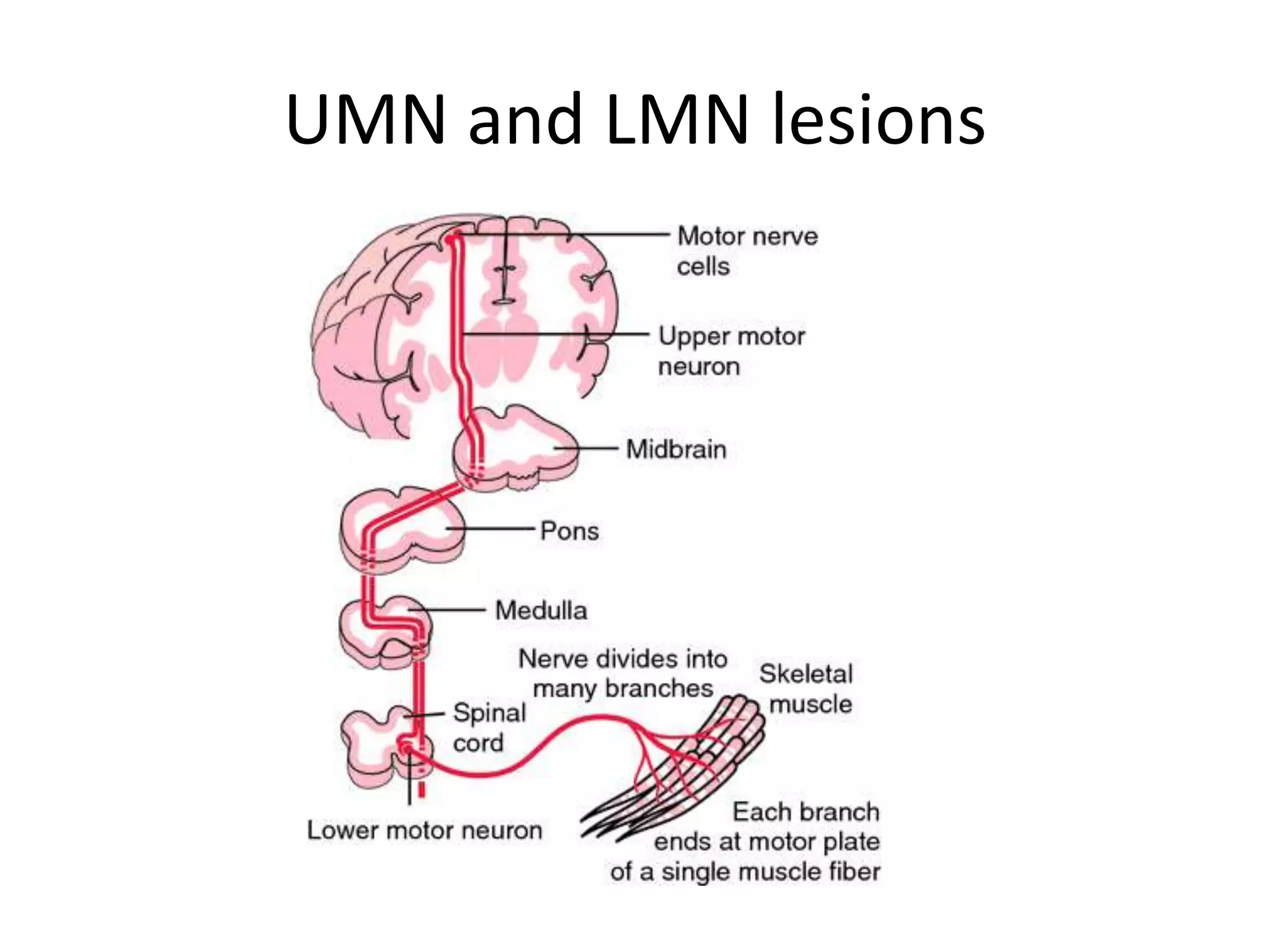
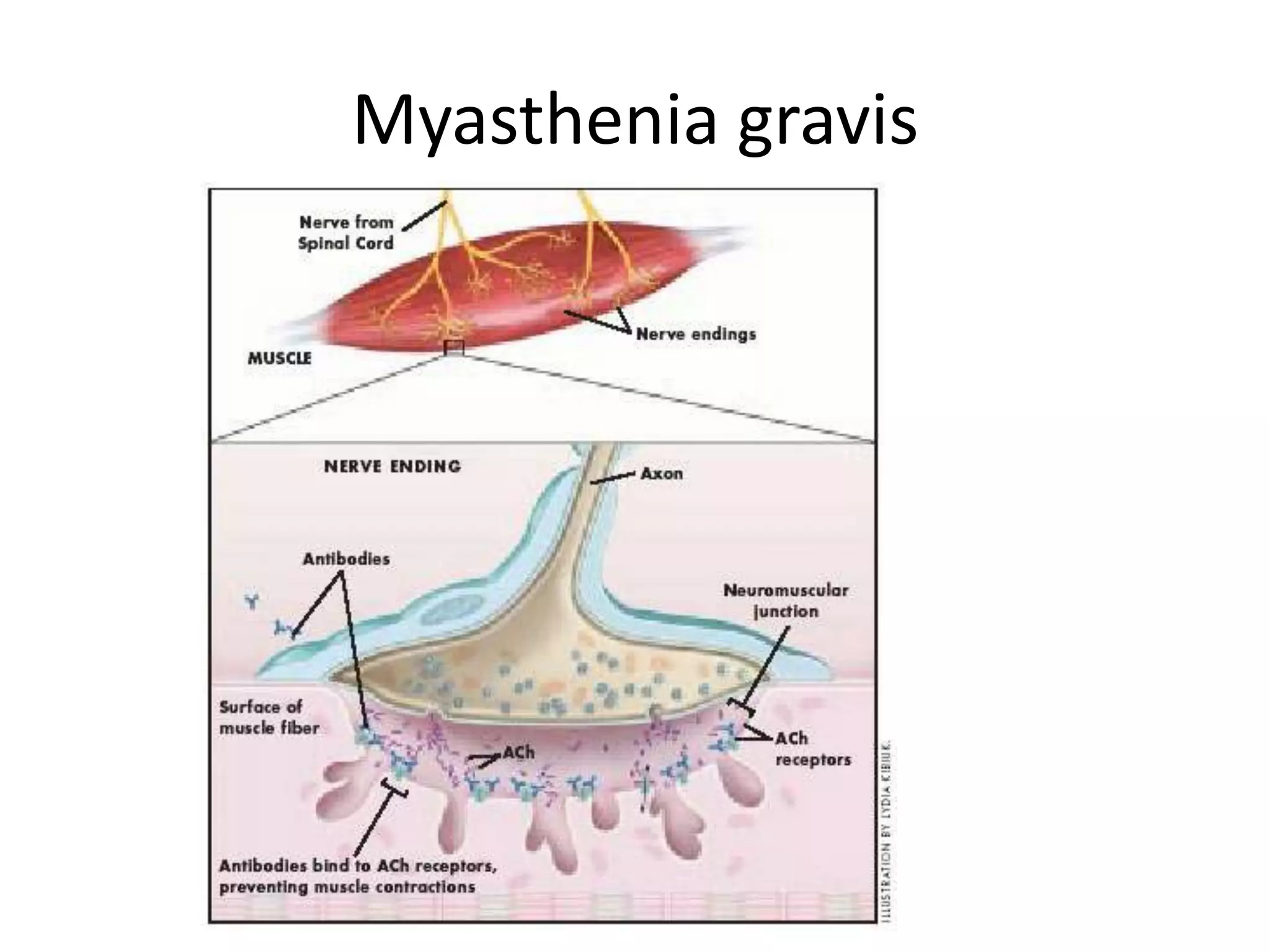


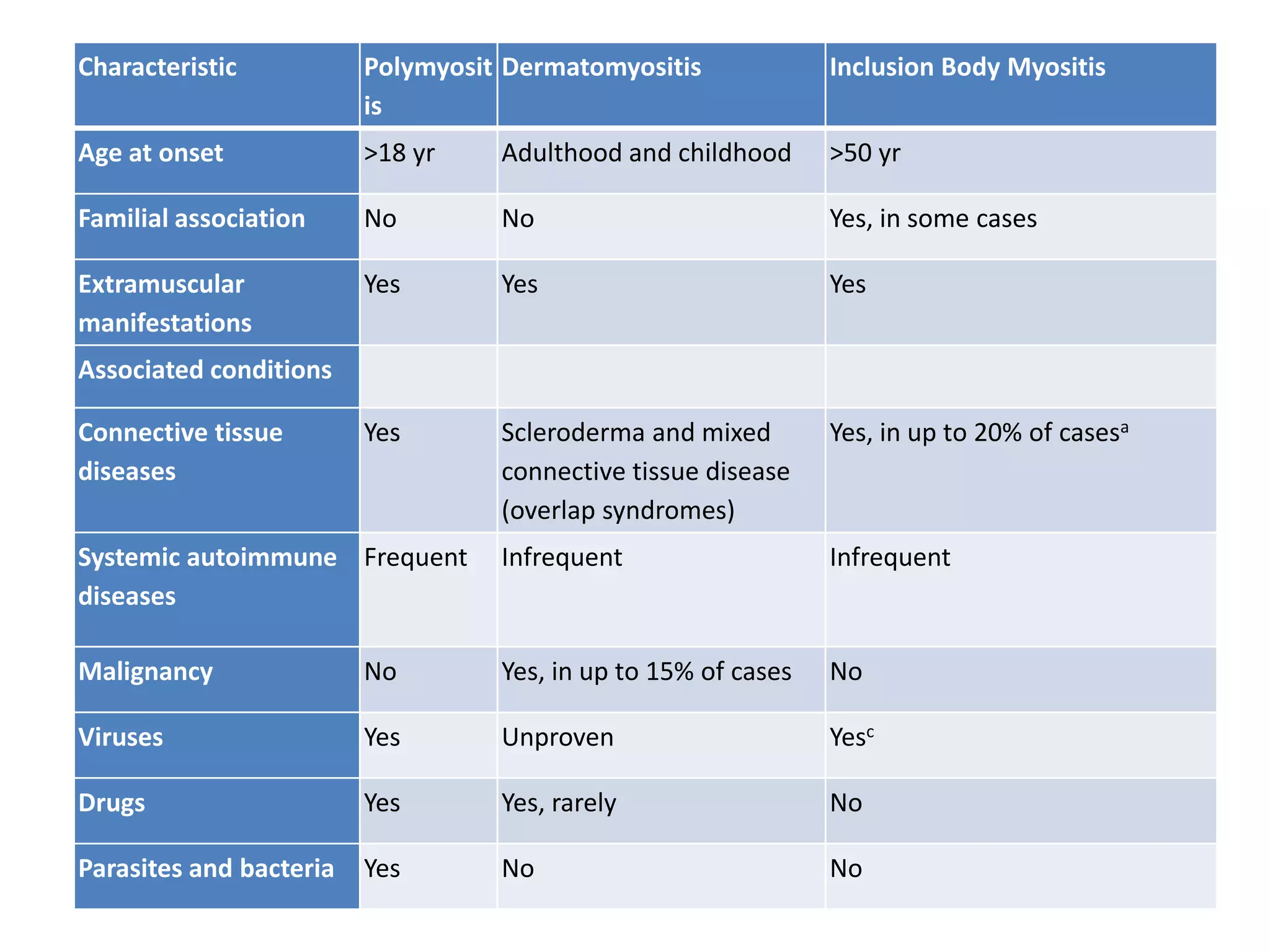




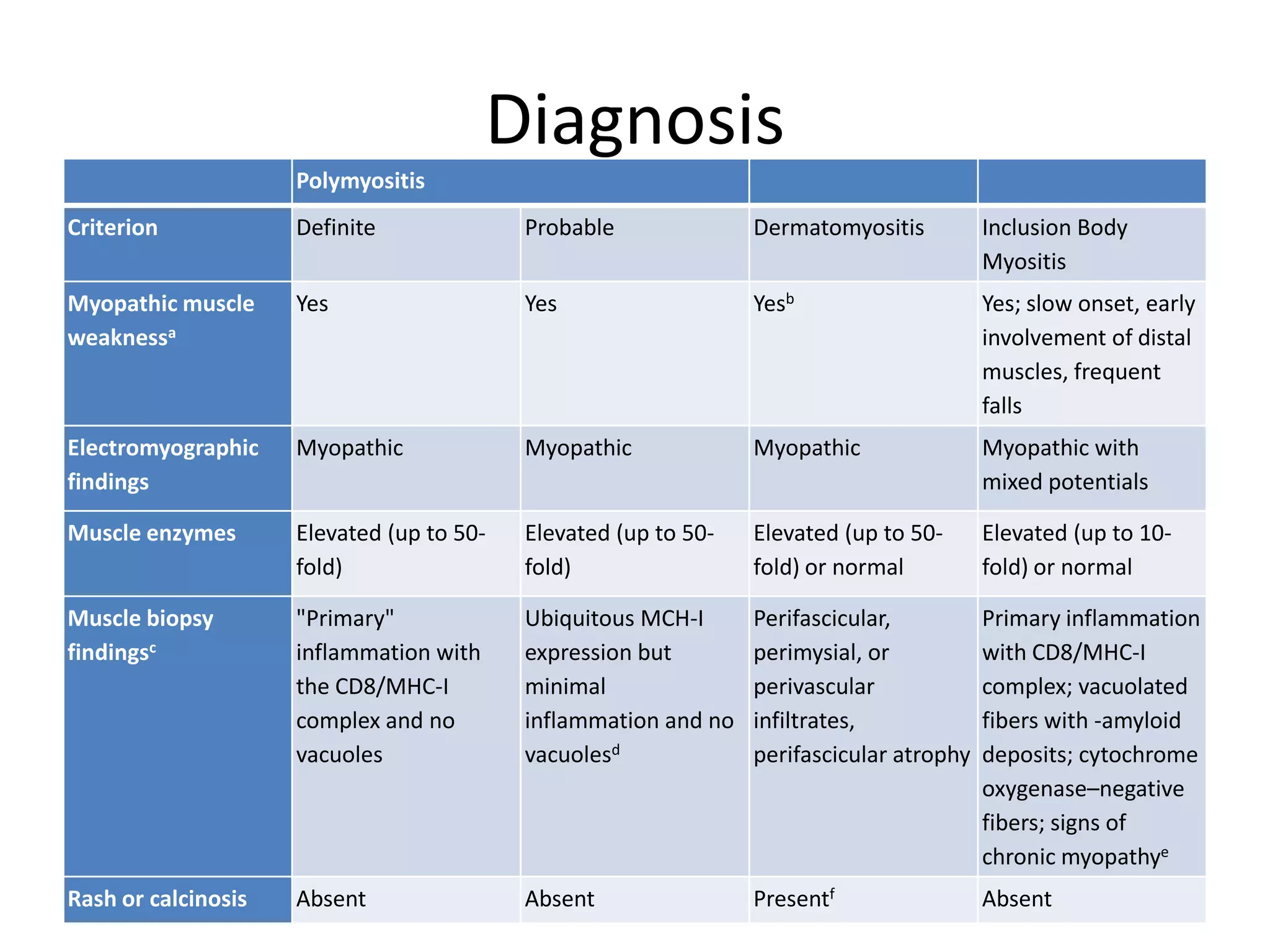



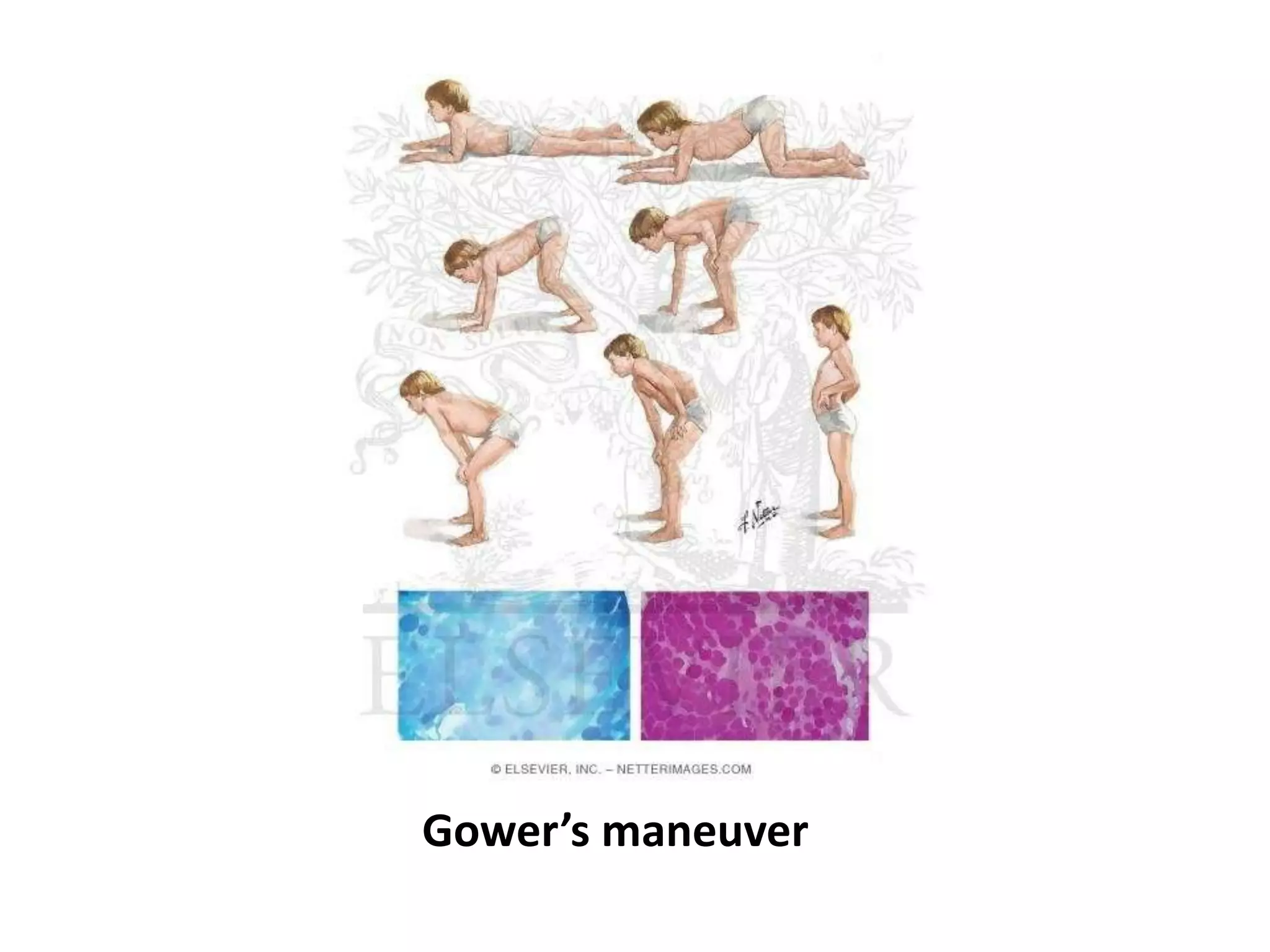




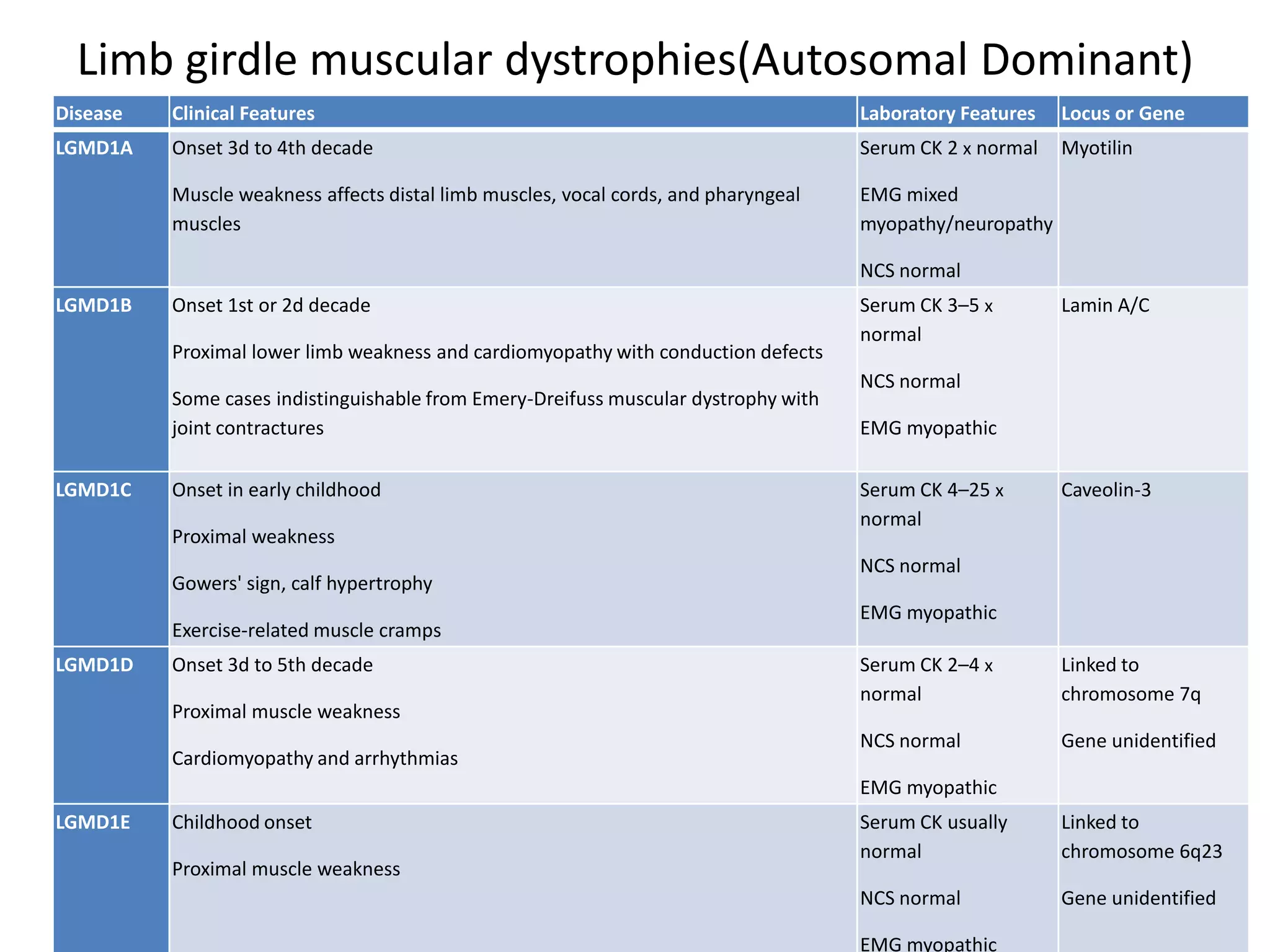
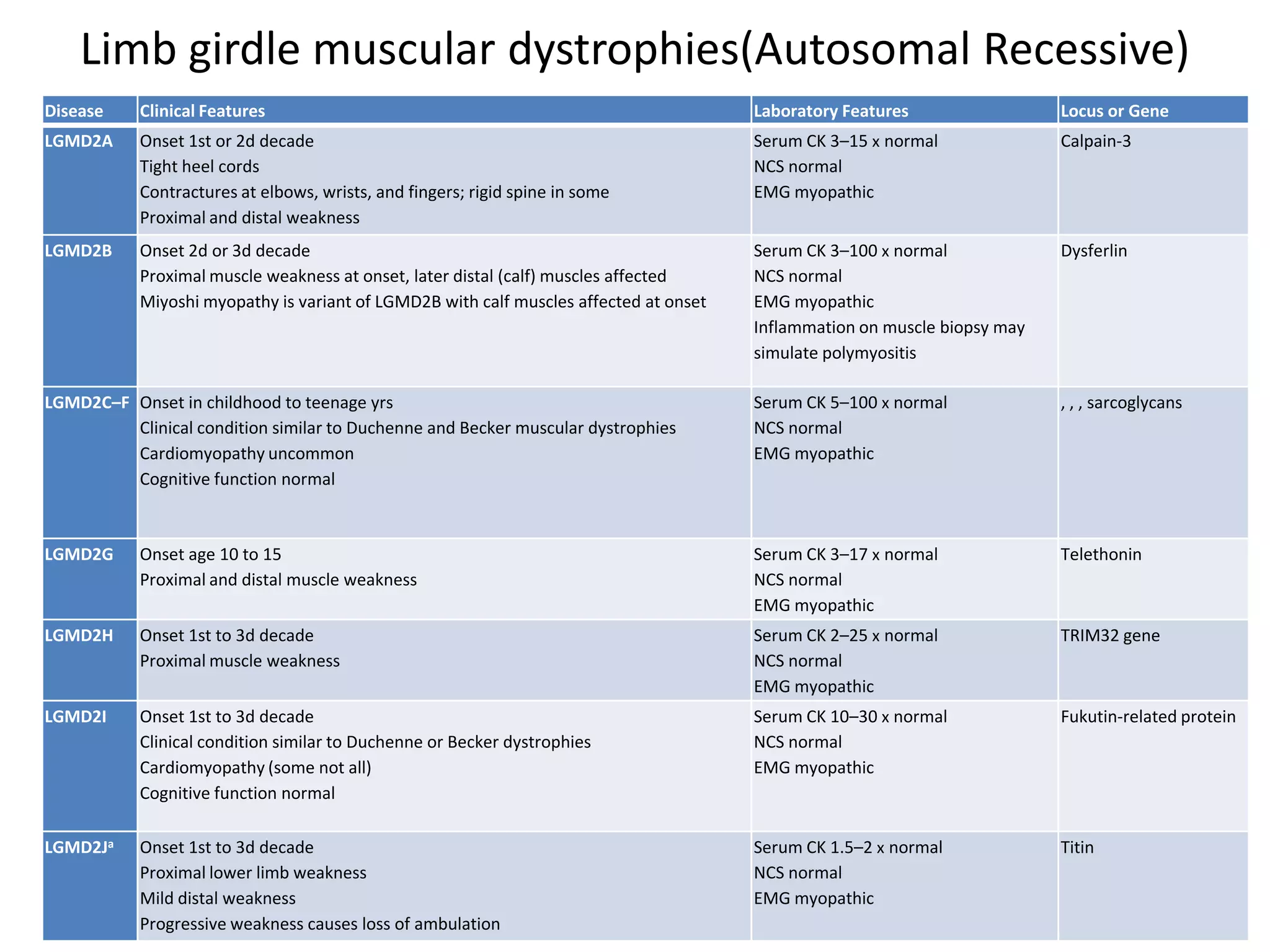
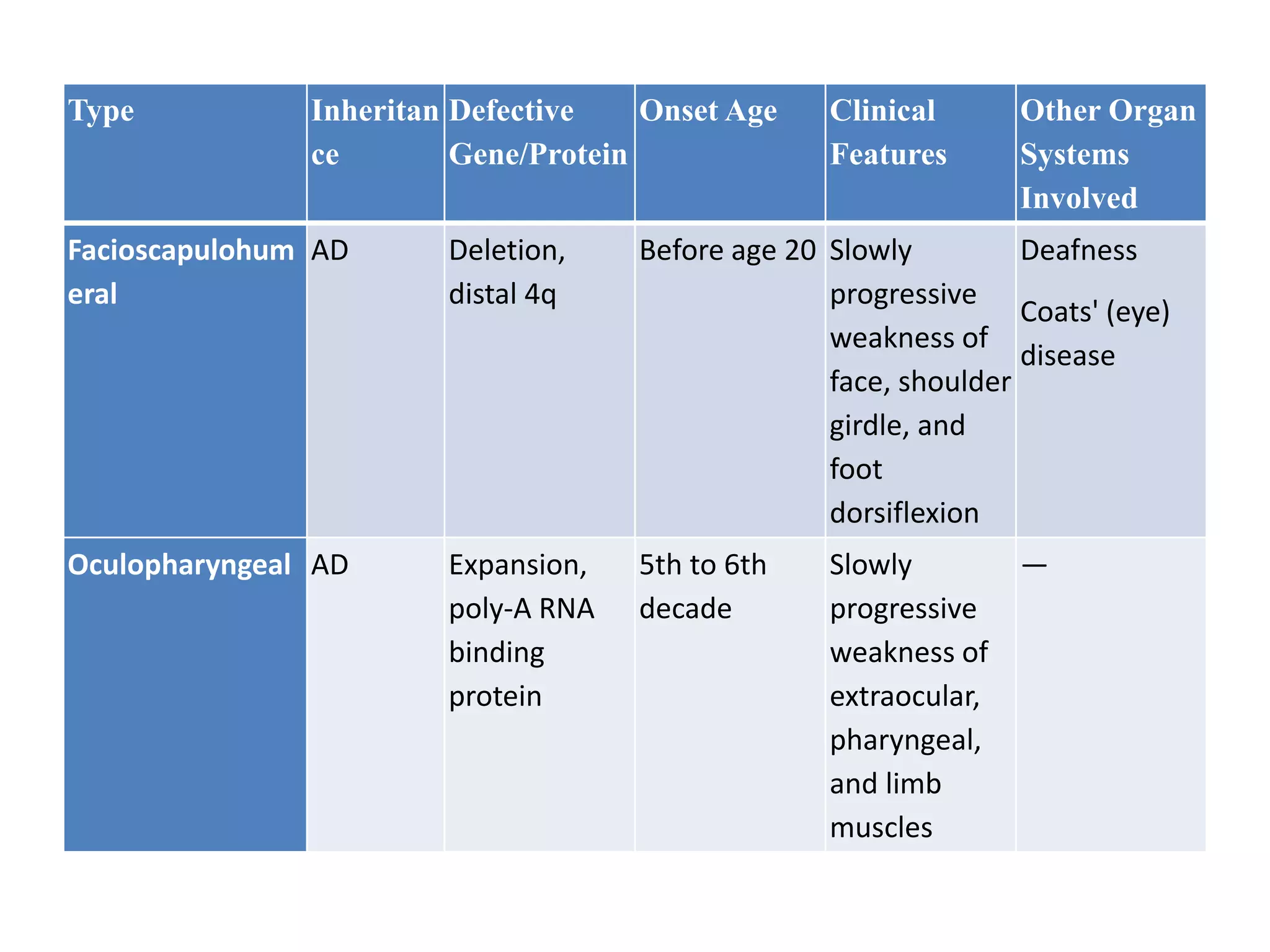
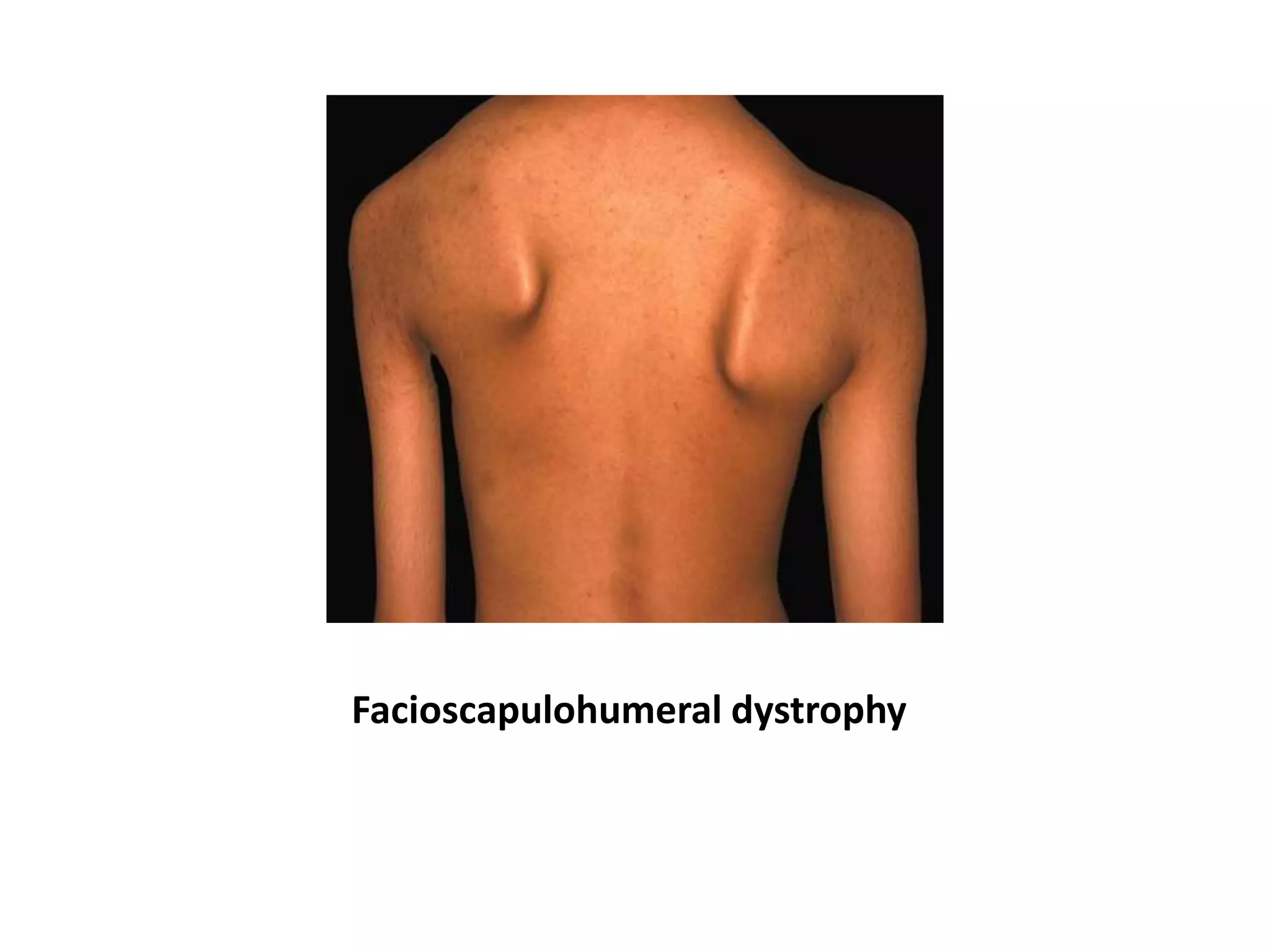


















This document discusses various types of myopathies (disorders affecting muscle). It defines myopathies and distinguishes them from other causes of muscle weakness. It then describes different categories of myopathies including inflammatory myopathies (such as polymyositis and dermatomyositis), muscular dystrophies (such as Duchenne, Becker, limb-girdle, facioscapulohumeral), congenital myopathies, metabolic myopathies, and others. For each type, it discusses inheritance, clinical features, diagnostic criteria, and treatment when available.























































































