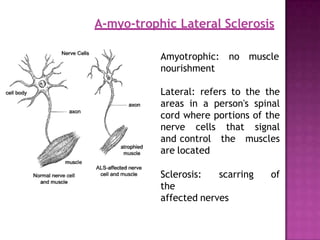
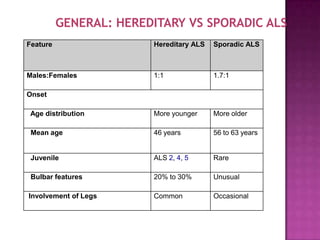
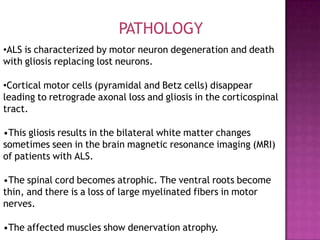
Motor neuron disease (MND) refers to a group of conditions characterized by degeneration of lower and/or upper motor neurons. Amyotrophic lateral sclerosis (ALS) is the most common form of MND, involving both upper and lower motor neurons. While Riluzole is the only FDA-approved treatment for ALS, other potential treatments discussed include ceftriaxone, tamoxifen, stem cell therapy, and supportive care approaches for symptoms such as spasticity, respiratory issues, and other complications.







![INCIDENCE AND PREV
ALENCE
•Incidence rates for ALS in Europe and North America range
between 1.5 and 2.7 per 100,000/year.[2]
•Prevalence rates range between 2.7 and 7.4 per 100,000 [2] .
•Incidence of ALS may be lower among African, Asian, and
Hispanic ethnic groups than among Caucasians [3].
•The male to female ratio is about 1.3 to 1.5 for sporadic ALS,
although the ratio becomes closer to unity in the age group
over 70 years.
•ALS is most commonly sporadic. Genetic or familial ALS
represents only 10 percent of all ALS
• In the United States, about 7000 new cases of ALS are
2. Worms, pm .the epidemiology of motor neuron disorder;a review of recent studies.j neuro sci 2001;191.
3. Cronin s, hardiman o,traynor BJ ethnic variation in the incidence of als ; a systemic review
.neurology 2007;68;1002.
diagnosed each year](https://image.slidesharecdn.com/bqpalz7sfiawafswxw5m-motor-neuron-disease-230528113347-06859980/85/Motor_neuron_disease-ppt-8-320.jpg)
![RISK FACTORS
•The only established risk factors for ALS are age and
family history.
•Increased risk for developing ALS has been suggested for
cigarette smokers, labourers engaged in agricultural work,
factory work, heavy manual labour, exposure to welding,
and work in the plastics industry .
•Repetitive muscle use, athleticism, playing professional
soccer, trauma, and electrical shock have also been
proposed as risk factors.
•A large case-control study found no association between
physical activity and the risk of developing ALS, but did find
that increased leisure time physical activity was associated
with a younger age of onset in patients with ALS [4].](https://image.slidesharecdn.com/bqpalz7sfiawafswxw5m-motor-neuron-disease-230528113347-06859980/85/Motor_neuron_disease-ppt-9-320.jpg)
![Excitotoxicity — The excitotoxicity hypothesis postulates
that excessive levels of the excitatory neurotransmitter
glutamate may initiate a cascade resulting in cellular death
of motor neurons in ALS.[9].
Excessive activation of glutamate receptors may lead to
increased entry of calcium into cells. In turn, intracellular
calcium may trigger a cascade of events that causes
neuronal cell death via
•Lipid peroxidation
•Nucleic acid damage
•and mitochondrial disruption.
9-VAN DEN BOSCH L , VAN DAMME P , BOGAERT E , ROBBERECHT W : The role of excitotoxicity in the pathogenesis
of amyotrophic lateral sclerosis. biochimica et Biophysica Acta ( 2006 ) 1762: 1068 - 1082](https://image.slidesharecdn.com/bqpalz7sfiawafswxw5m-motor-neuron-disease-230528113347-06859980/85/Motor_neuron_disease-ppt-12-320.jpg)
![Apoptosis or programmed cell death cascades have been
implicated in several studies .
These reports have shown a number of the hallmarks of
apoptosis including
•DNA fragmentation
•altered expression of the antiapoptotic protein Bcl-2 [16]
Apoptosis
16-LI M , ONA VO , GUEGAN C et al. : Functional role of caspase 1 and 3 in ALS. Science ( 2000 ) 288 :335 - 339](https://image.slidesharecdn.com/bqpalz7sfiawafswxw5m-motor-neuron-disease-230528113347-06859980/85/Motor_neuron_disease-ppt-13-320.jpg)









































































































