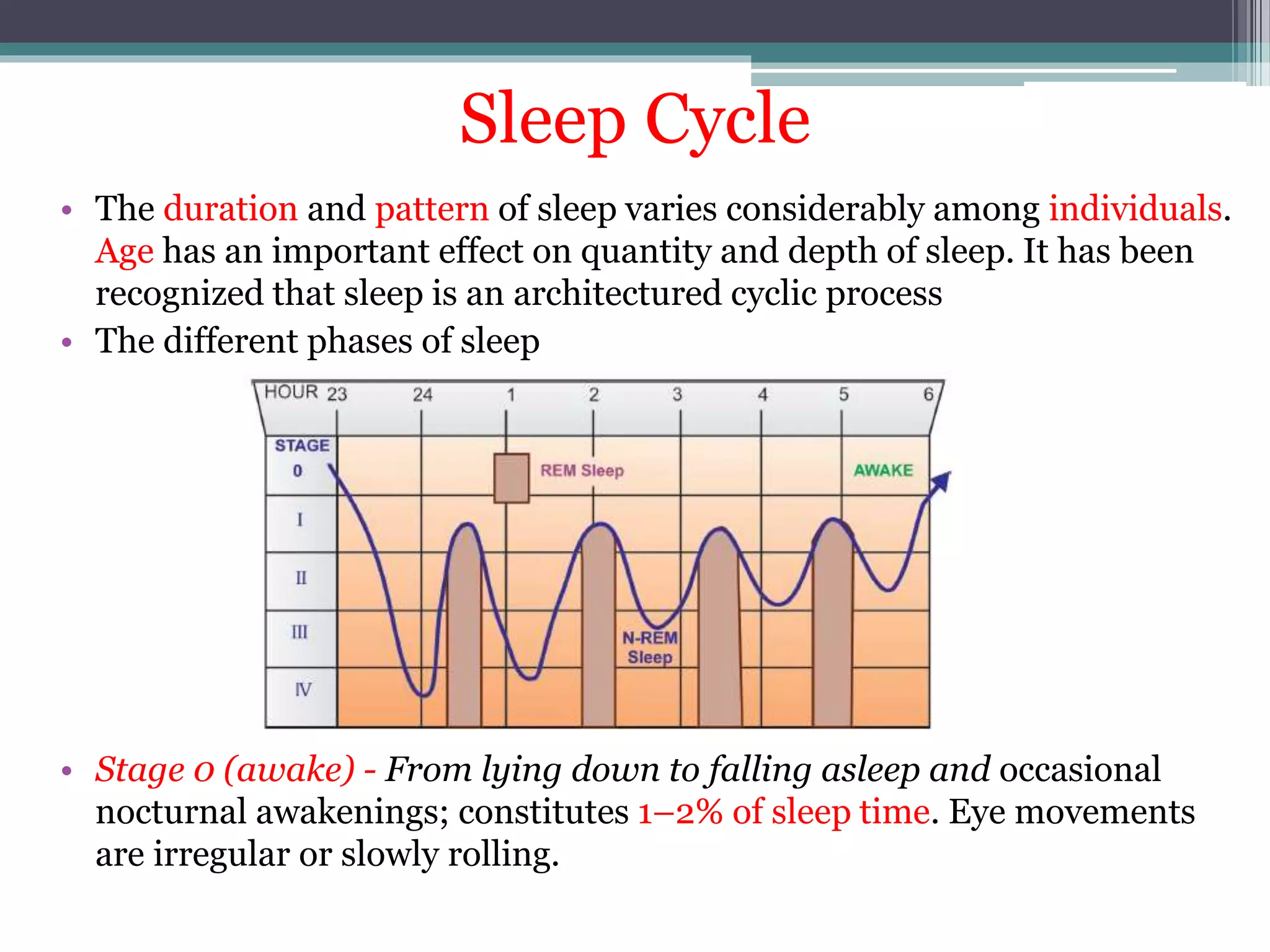
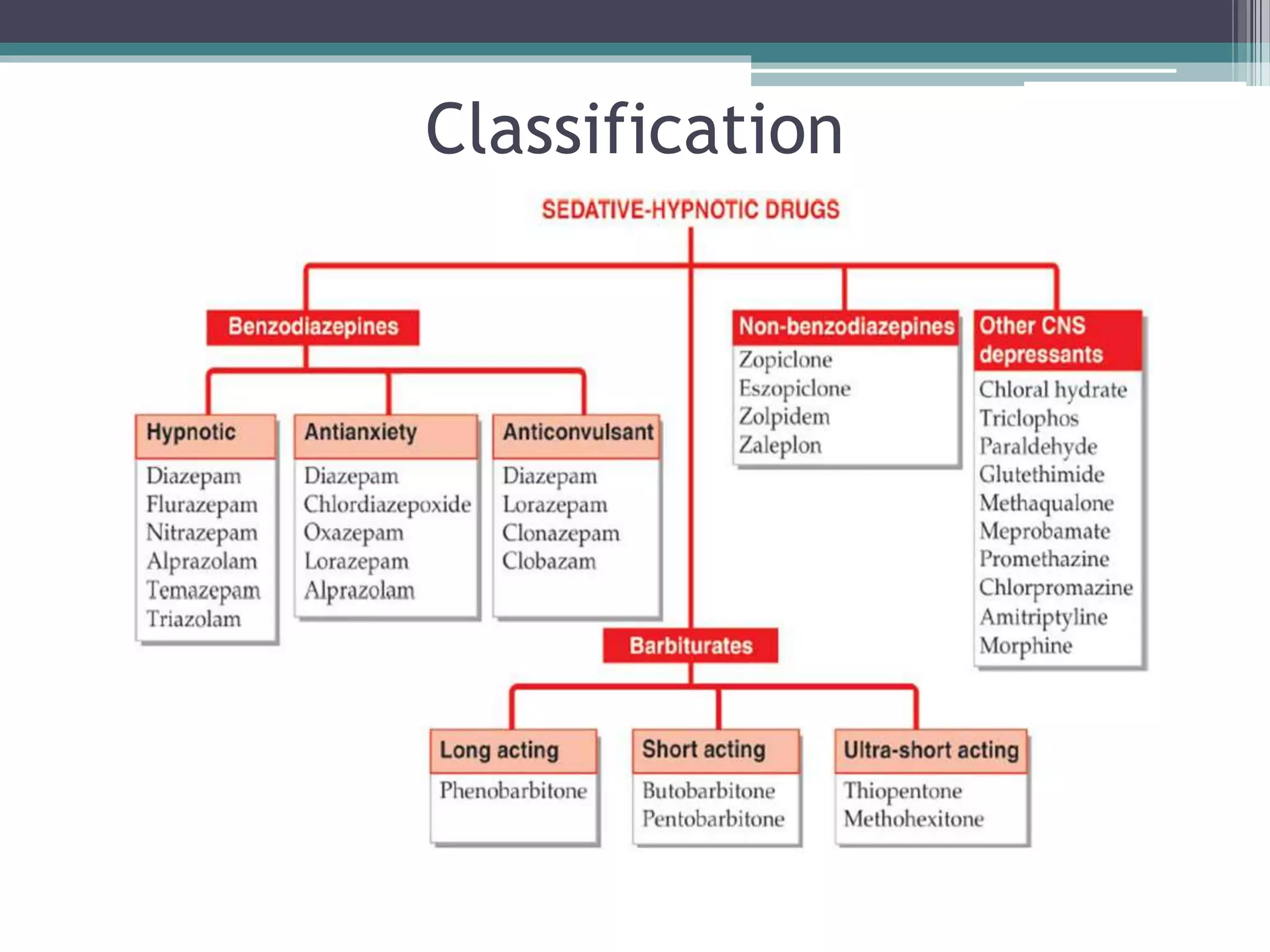
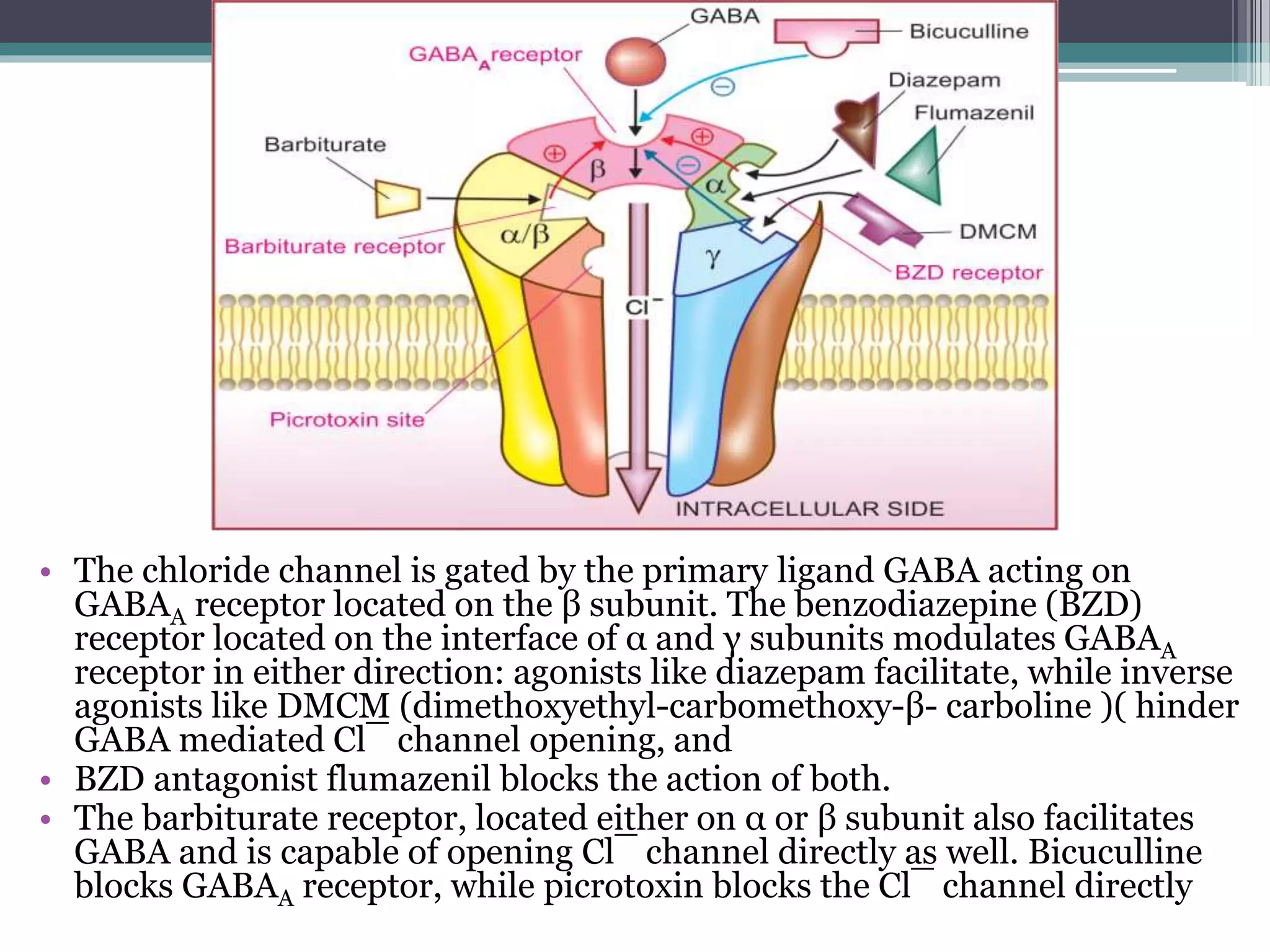
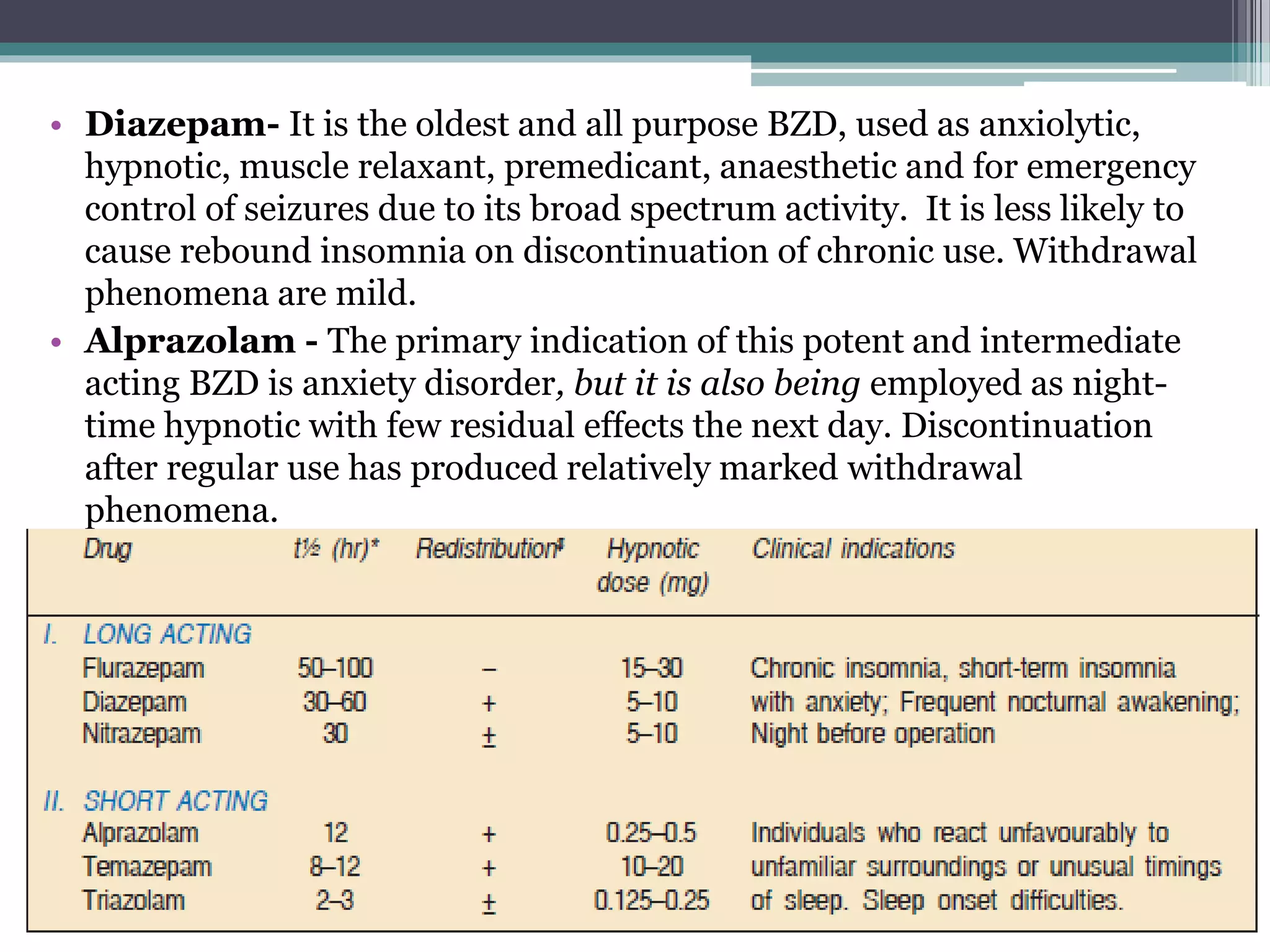
This document discusses sedative-hypnotics, including their classification, mechanisms of action, and uses. It covers barbiturates, benzodiazepines, and non-benzodiazepine hypnotics. Barbiturates were formerly used as hypnotics and sedatives but have mostly been replaced by benzodiazepines due to lower toxicity. Both barbiturates and benzodiazepines act by enhancing the effects of the inhibitory neurotransmitter GABA. Non-benzodiazepine hypnotics act through specific benzodiazepine receptors to induce sleep with fewer side effects than benzodiazepines. Hypnotics are used for short-term