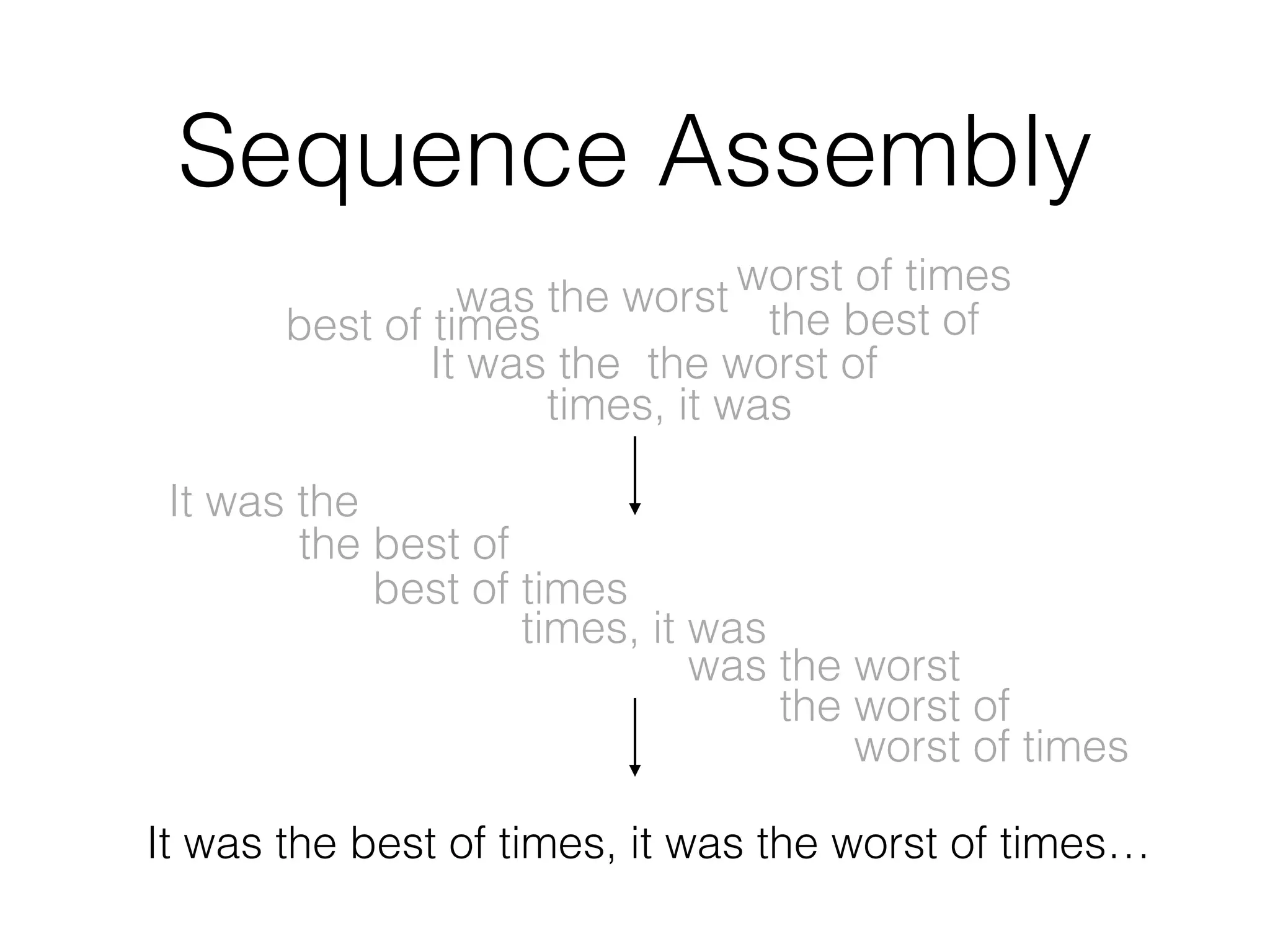
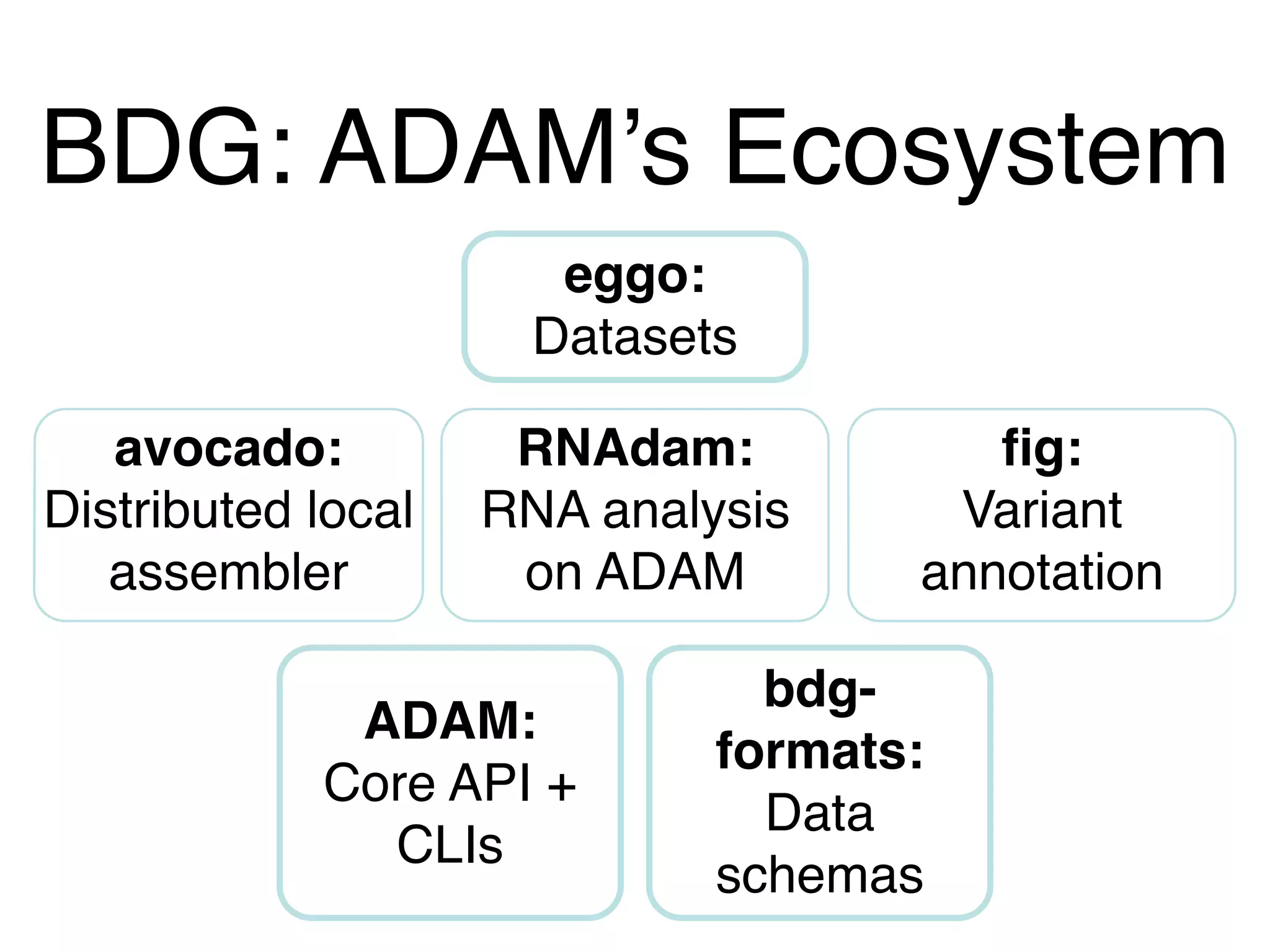
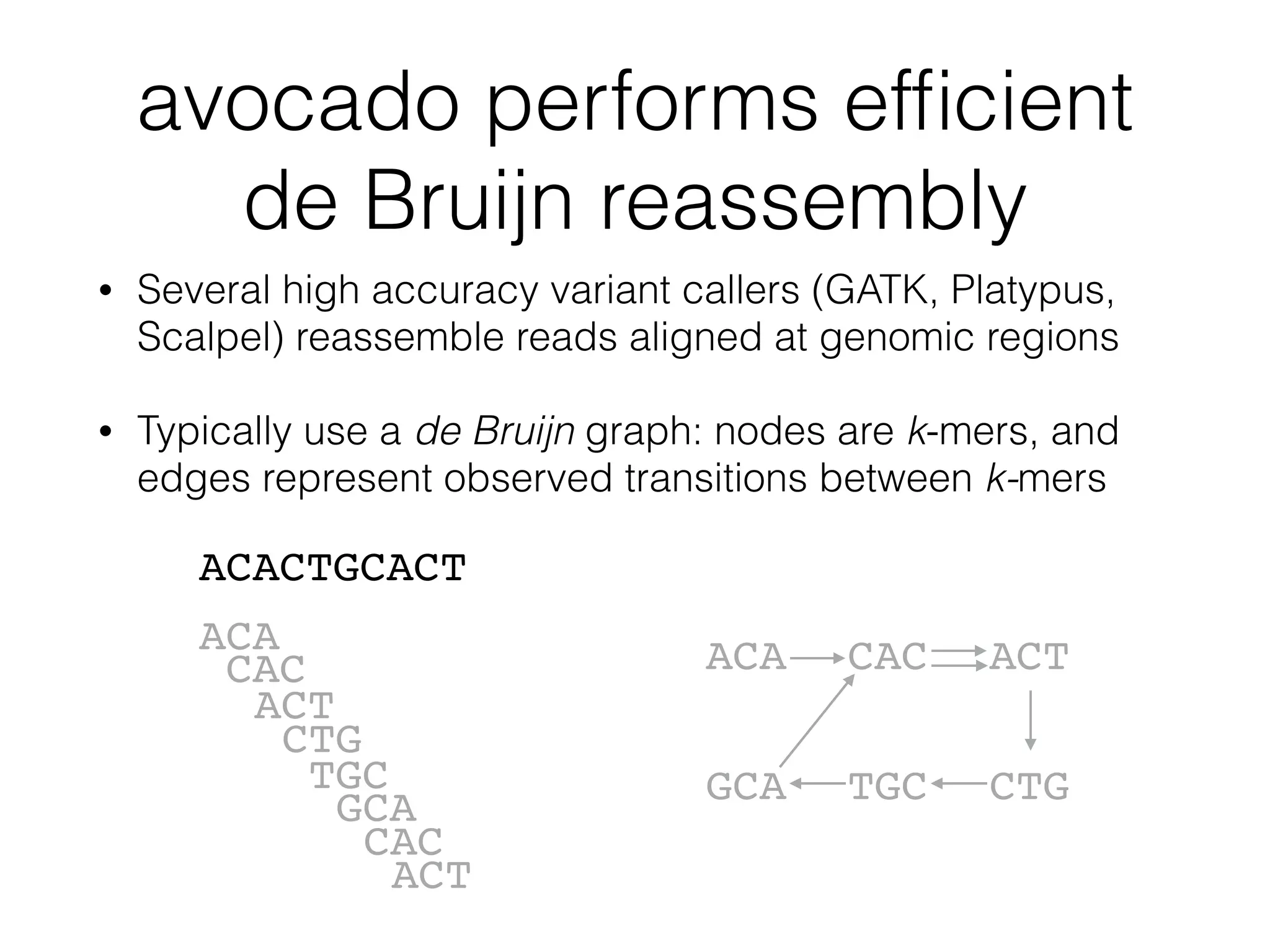
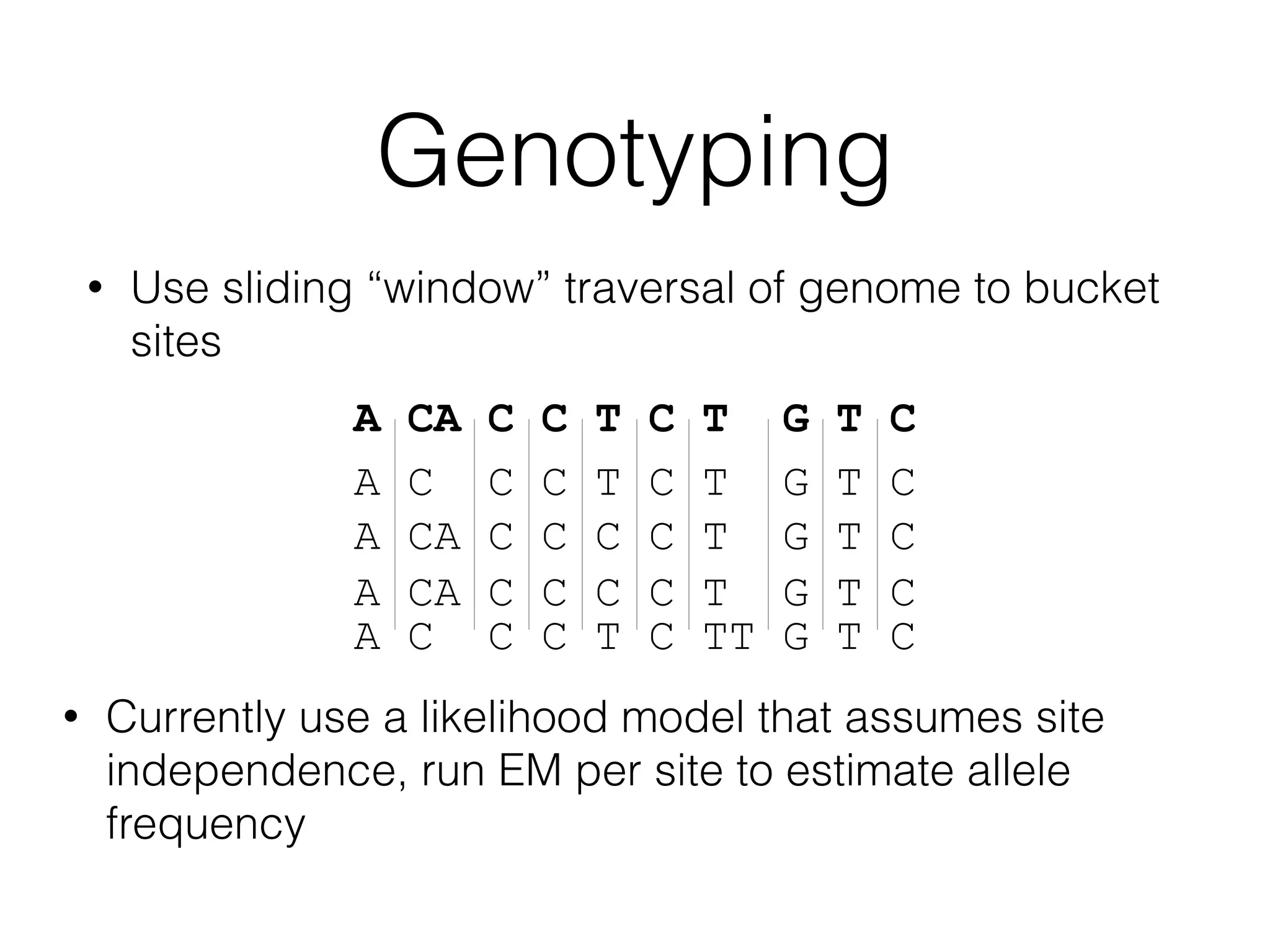
This document provides a summary of the Scalable Genome Analysis with ADAM project. ADAM is an open-source, high-performance, distributed platform for genomic analysis that defines a data schema, data layout on disk, and programming interface for distributed processing of genomic data using Spark and Scala. The goal of ADAM is to integrate across terabyte and petabyte-scale datasets to enable the discovery of low frequency genetic variants linked to traits and diseases.