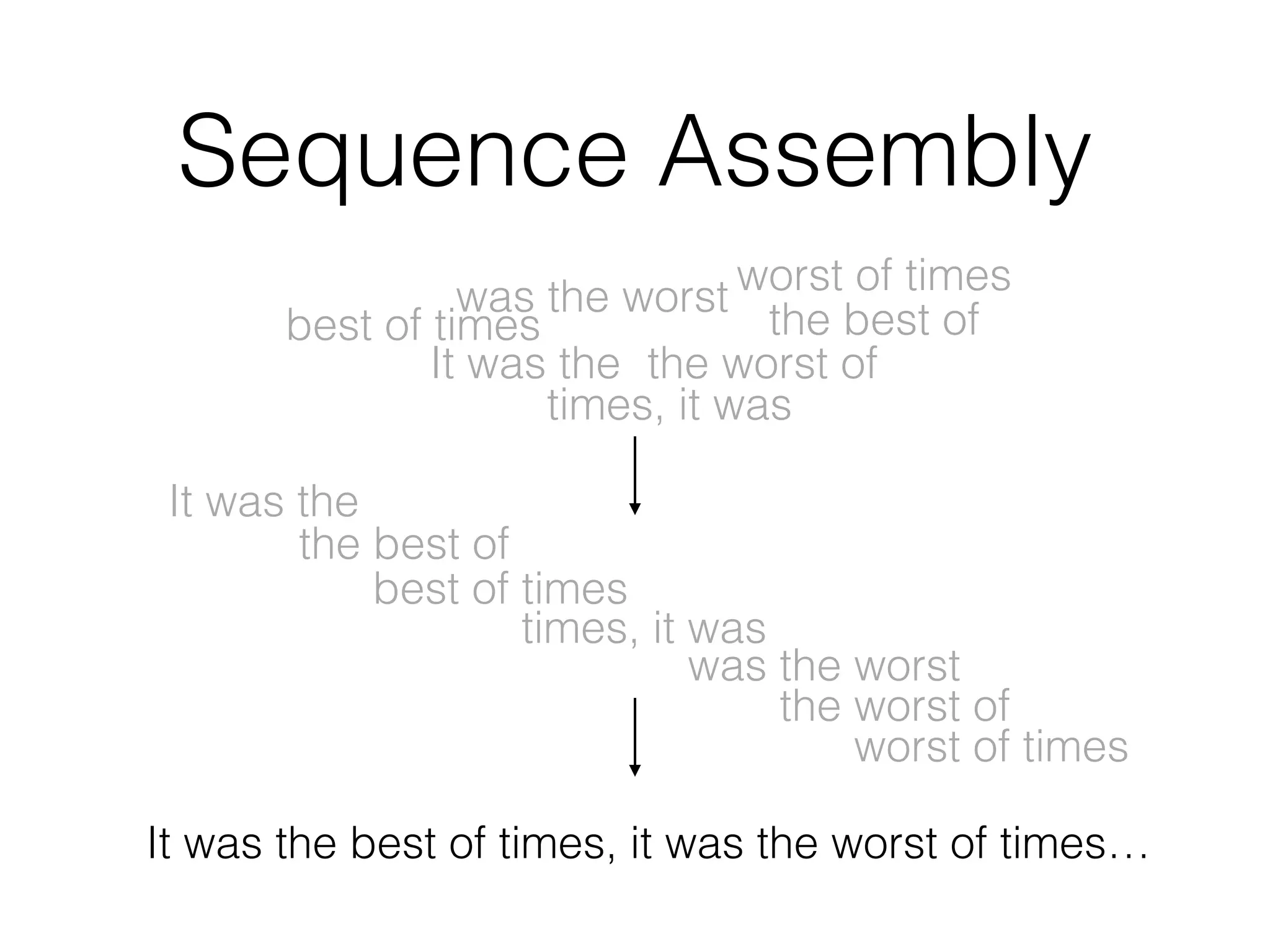
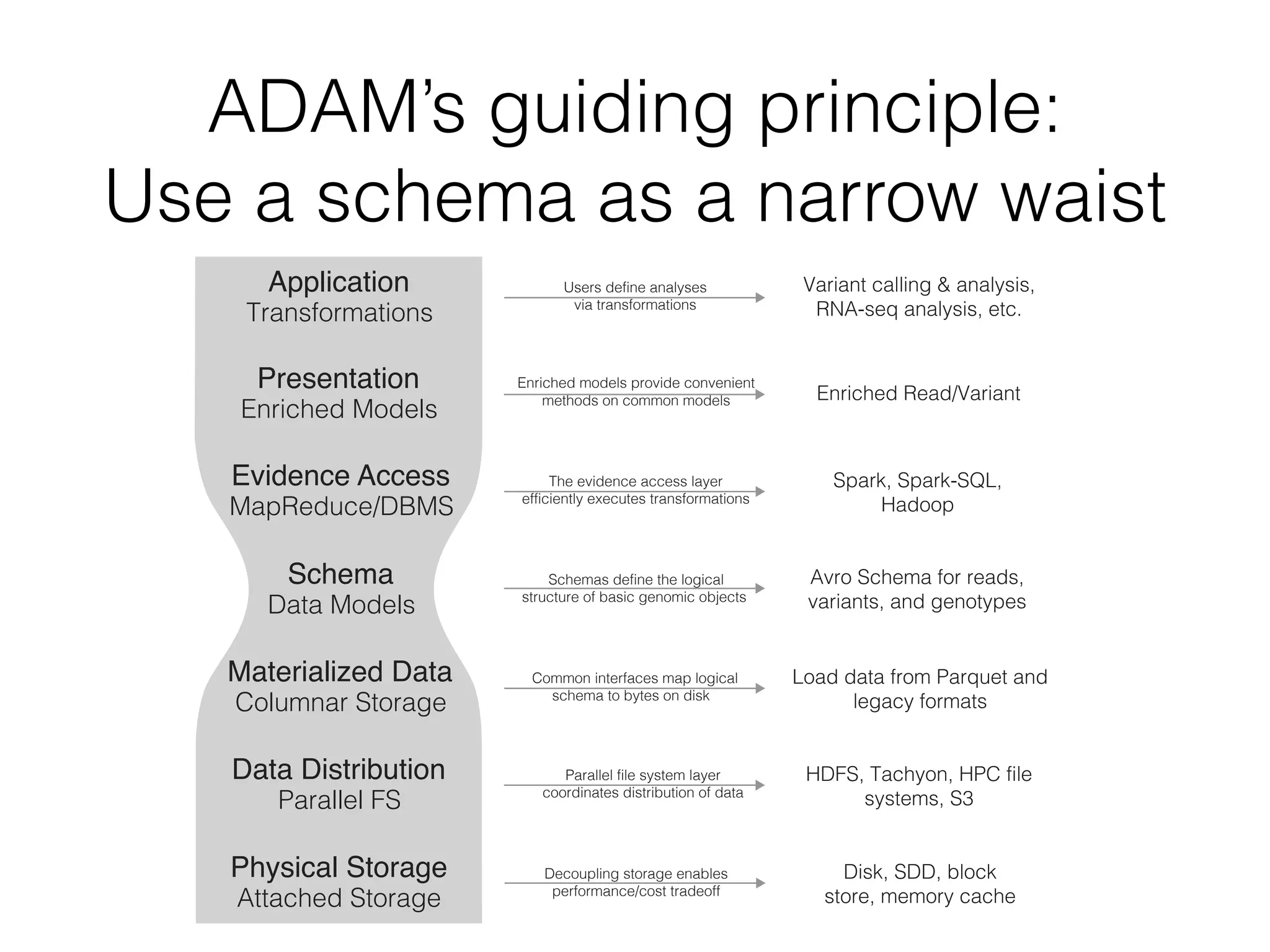
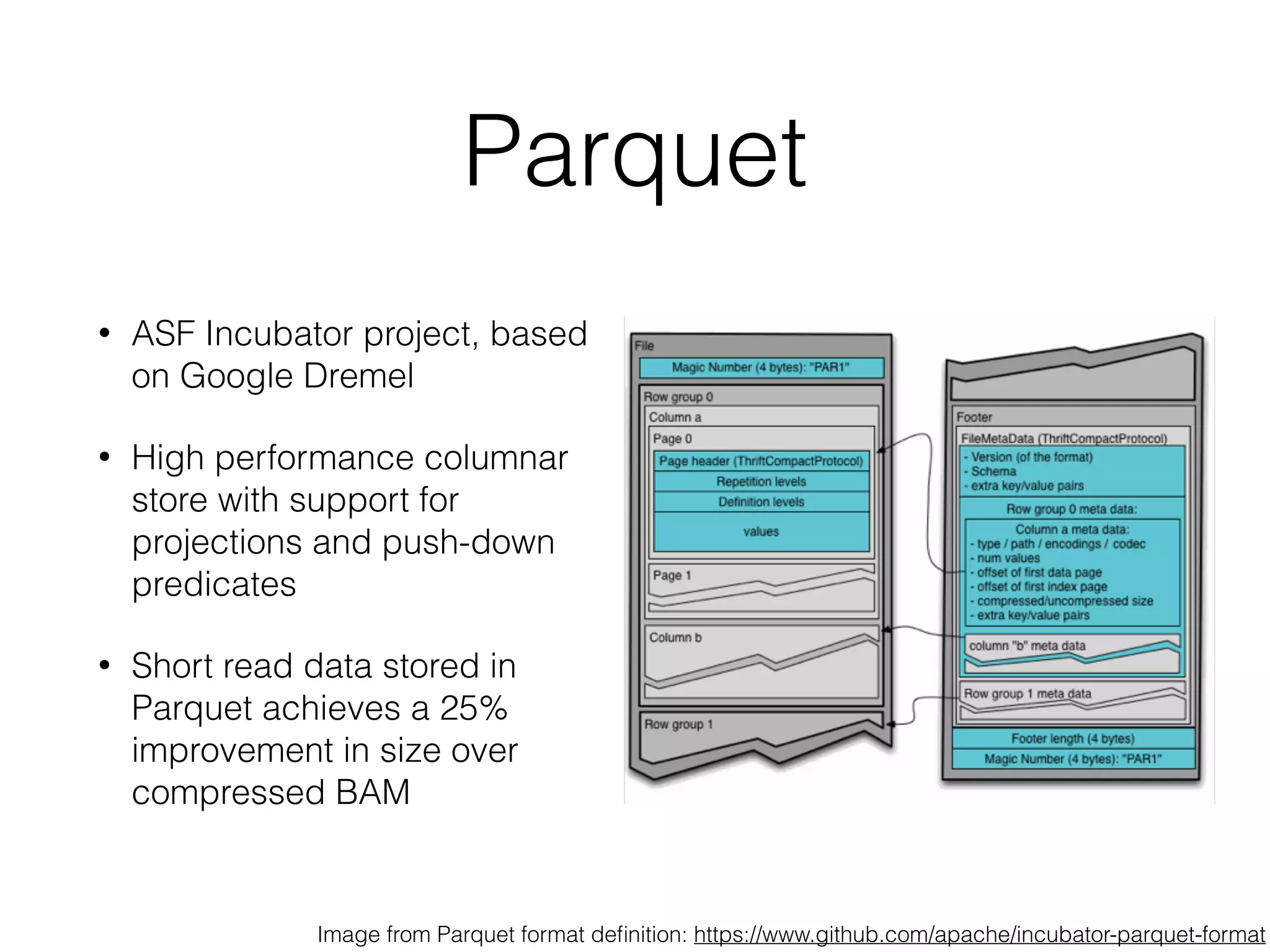
This document discusses scalable genome analysis using ADAM (Apache Spark-based framework). It begins by describing genomes and the goal of analyzing genetic variations. The document then discusses challenges like the large size of genomes and complexity of linking variations to traits. It proposes using ADAM's schema, optimized storage and algorithms to accelerate common access patterns like overlap joins. The document also emphasizes applying biological knowledge like protein grammars to make sense of non-coding variations. Finally, it acknowledges contributions from various institutions that have helped develop ADAM and its ability to enable genome analysis at scale.