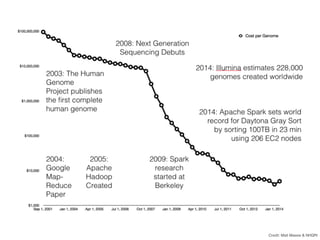
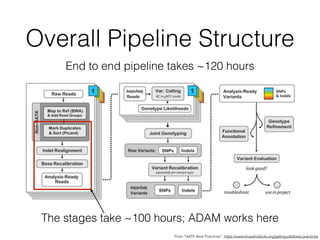
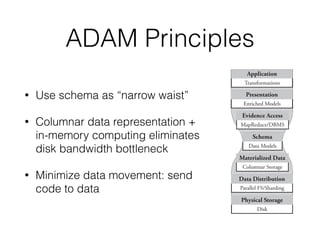
The document discusses ADAM, a new framework for scalable genomic data analysis. It aims to make genomic pipelines horizontally scalable by using a columnar data format and in-memory computing. This avoids disk I/O bottlenecks. The framework represents genomic data as schemas and stores data in Parquet for efficient column-based access. It has been shown to reduce genome analysis pipeline times from 100 hours to 1 hour by enabling analysis on large datasets in parallel across many nodes.