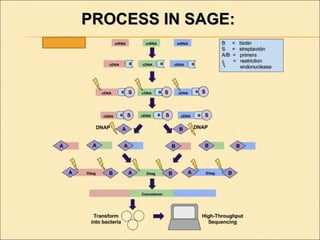
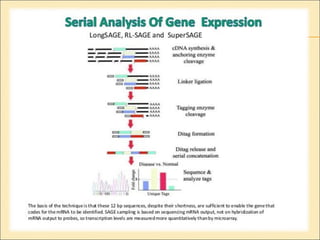
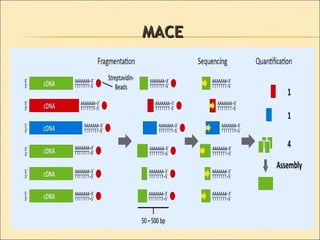
Serial Analysis of Gene Expression (SAGE) is a transcriptomic technique that generates a snapshot of mRNA populations using small tags corresponding to RNA fragments. The process involves isolating mRNA, synthesizing cDNA, and creating concatemers of tagged fragments for sequencing, allowing for detailed analysis of gene expression. Variants of the original technique, such as LongSAGE, SuperSAGE, and MACE, have been developed to improve throughput, precision, and the ability to analyze non-model organisms.