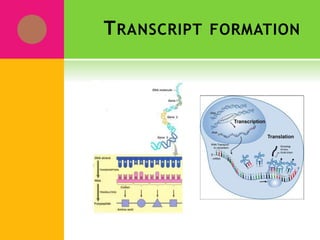
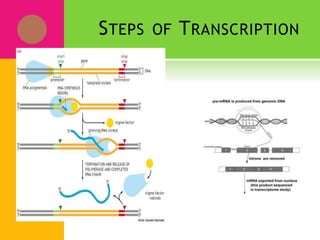
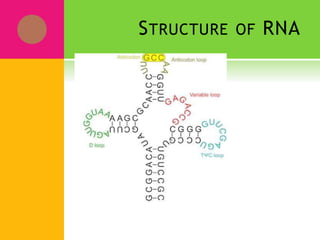
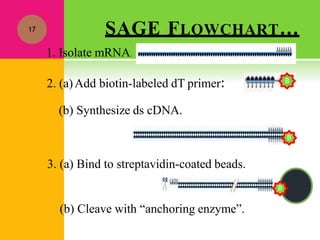
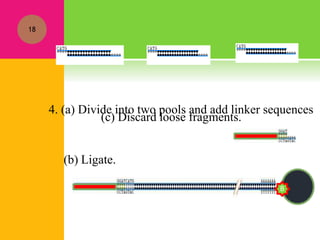
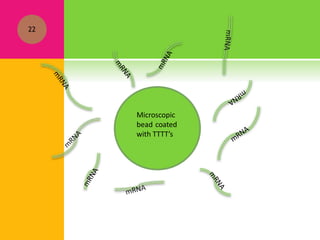
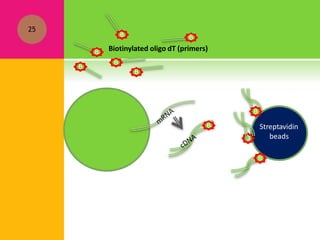
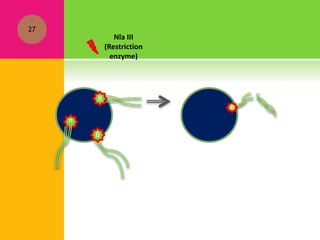
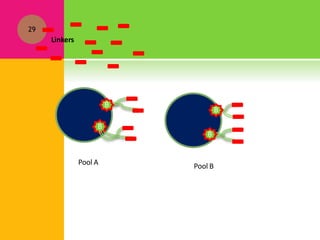
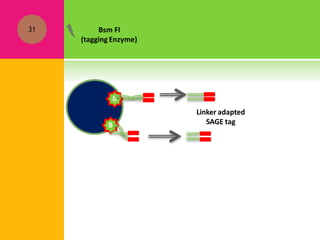
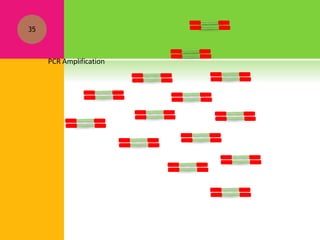
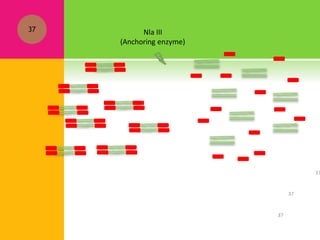
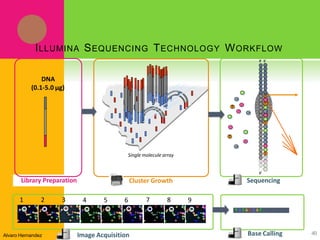
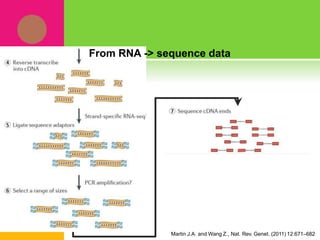
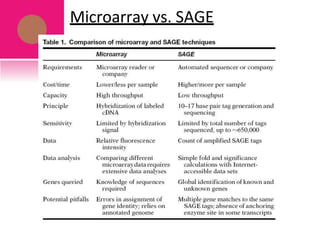
The document provides a history of transcriptomics and an overview of RNA analysis techniques. It begins by explaining Francis Crick's central dogma and the discovery of messenger RNA, transfer RNA, and ribosomal RNA in the 1960s. Later developments include the discoveries of RNA splicing, ribozymes, RNA interference, and small interfering RNA. The document then defines the transcriptome and describes methods for analyzing RNA expression like transcript formation, RNA structure, positional integration on the transcriptome, Northern blotting, serial analysis of gene expression (SAGE), and the basic steps of SAGE.