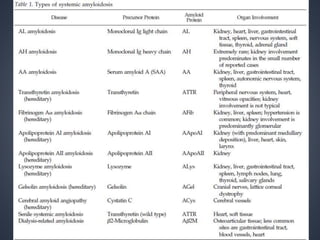
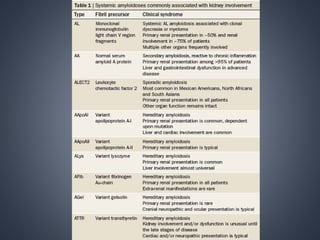
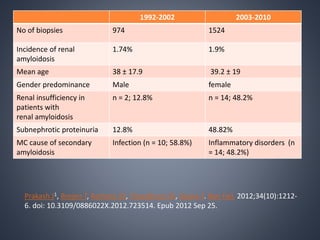
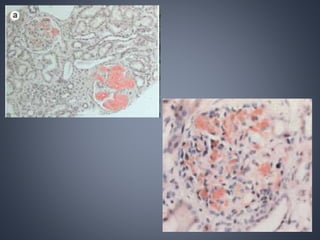
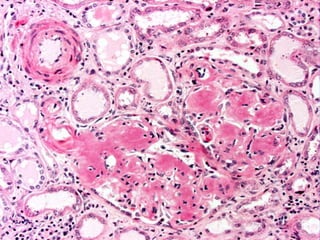
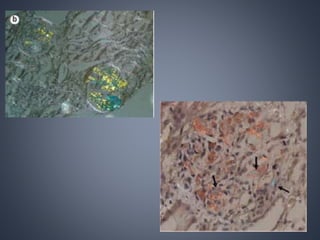
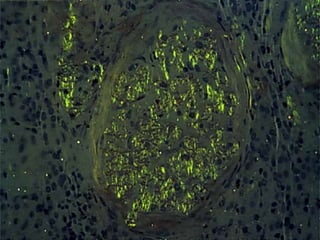
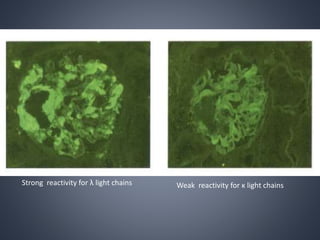
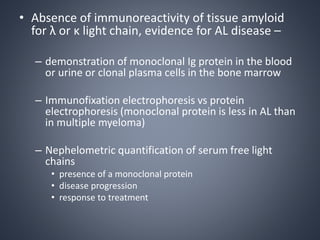
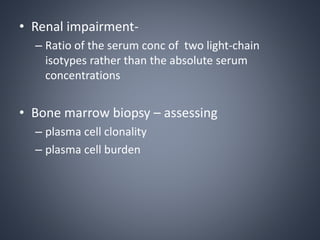
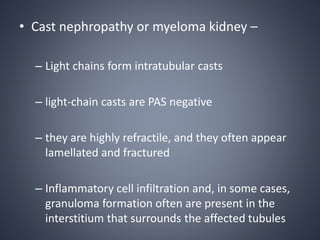
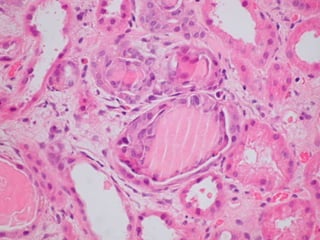
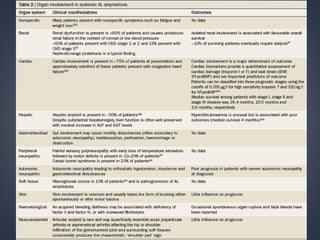
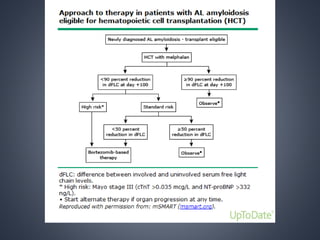
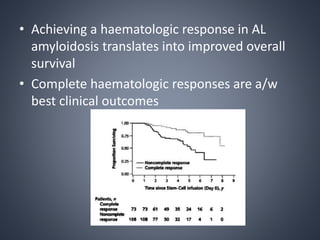
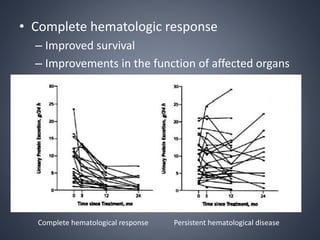
This document provides a review of renal amyloidosis. It begins by defining amyloidosis as a group of diseases caused by the misfolding and accumulation of various proteins. 27 human proteins are known to cause amyloidosis. The kidney is a common site of deposition for several types of amyloidosis. The document reviews the pathogenesis of amyloidosis, determinants of renal deposition, how it causes renal disease, classification, epidemiology including statistics from India, pathology findings including staining techniques, and methods to determine the type of amyloidosis involved.




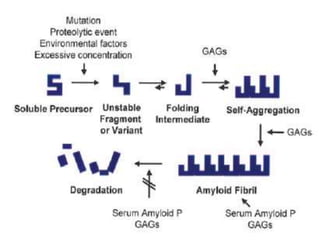
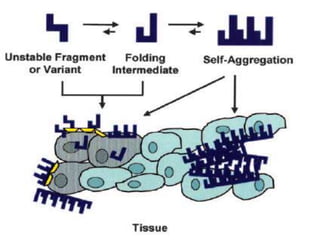
![Pathogenesis of amyloidosis
• For each of these amyloidogenic “precursor proteins,”
– initial step in amyloid fibril formation is a misfolding event
• Misfolding can result from
– Proteolytic cleavage (e.g., amyloid β protein)
– An amino acid substitution (e.g., transthyretin [TTR])
– Intrinsic properties that become significant only at high
serum concentration or in the presence of specific local
factors (e.g., β2-microglobulin)](https://image.slidesharecdn.com/renalamyloidosis-150513070906-lva1-app6891/85/Renal-amyloidosis-4-320.jpg)